
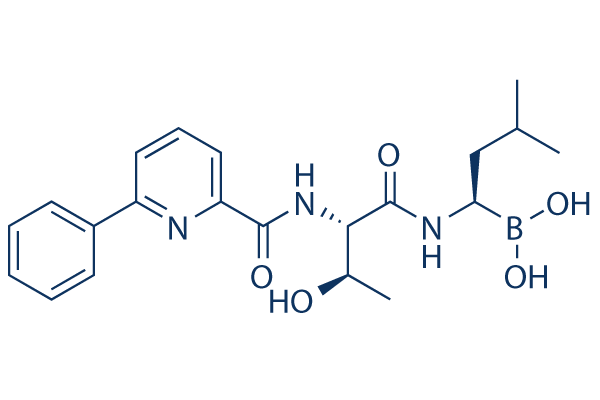
Tazemetostat
Current developer: Epizyme, Inc., Cambridge, MA 02139.
EPZ-6438 (Tazemetostat)
CAS: 1403254-99-8
Chemical Formula: C34H44N4O4
Exact Mass: 572.33626
N-((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[1,1′-biphenyl]-3-carboxamide
SIMLES: O=C(C1=CC(C2=CC=C(CN3CCOCC3)C=C2)=CC(N(CC)C4CCOCC4)=C1C)NCC5=C(C)C=C(C)NC5=O
(1,1′-Biphenyl)-3-carboxamide, N-((1,2-dihydro-4,6-dimethyl-2-oxo-3-pyridinyl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(4-morpholinylmethyl)-
N-((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-5-(ethyl(oxan-4-yl)amino)-4-methyl-4′-((morpholin-4-yl)methyl)(1,1′-biphenyl)-3-carboxamide
UNII-Q40W93WPE1
WO 2012142504 PRODUCT PAT
SEE Proceedings of the National Academy of Sciences of the United States of America (2013), 110(19), 7922-7927, S7922/1-S7922/5….http://www.pnas.org/content/110/19/7922.abstract
http://www.epizyme.com/wp-content/uploads/2014/11/Ribrag-ENA-FINAL.pdf
Tazemetostat, also known as EPZ-6438, is a potent, selective, and orally bioavailable small-molecule inhibitor of EZH2 enzymatic activity. EPZ-6438 induces apoptosis and differentiation specifically in SMARCB1-deleted MRT cells.
Treatment of xenograft-bearing mice with EPZ-6438 leads to dose-dependent regression of MRTs with correlative diminution of intratumoral trimethylation levels of lysine 27 on histone H3, and prevention of tumor regrowth after dosing cessation.
These data demonstrate the dependency of SMARCB1 mutant MRTs on EZH2 enzymatic activity and portend the utility of EZH2-targeted drugs for the treatment of these genetically defined cancers. EPZ-6438 is currently in clinical trials.
Epizyme, Inc., Eisai R&D Management Co.Ltd.
Epizyme is developing tazemetostat, a lead from several small molecule EZH2 inhibitors, for treating cancer (phase 1 clinical, as of April 2015). Japanese licensee Eisai was developing the program for the potential oral treatment of cancers, including non-Hodgkin’s lymphoma; however, in March 2015, Epizyme regained worldwide, ex-Japan, rights to the program.
It appeared that Eisai was planning to investigate the program in Japan .
WO-2015057859 From, Eisai Research Institute; Epizyme Inc, indicates Novel crystalline polymorphic form C of tazemetostat, useful for treating an EZH2-mediated cancer, including non-Hodgkin’s lymphoma and breast cancer.
see WO2013155317, claiming novel hydrobromide salt of tazemetostat.
PREDICT



………………………………….
PATENT
WO 2012142504
http://www.google.com/patents/WO2012142504A1?cl=en

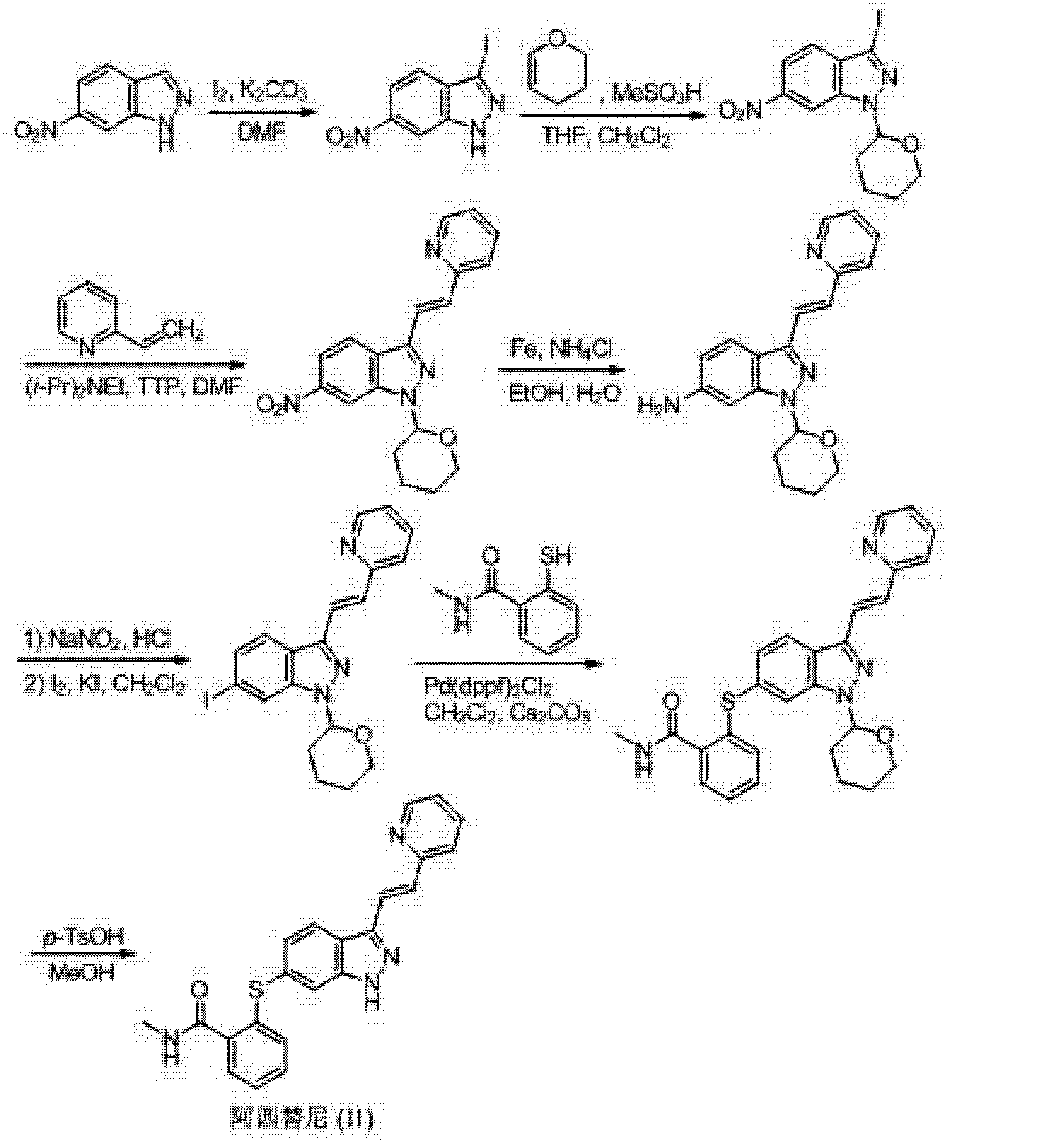
Example 44: Synthesis of N-((4,6-dimethyl-2-oxo-l ,2-dihydropyridin-3- yl)methyl)-5-(ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(moφholinomethyl)-[l , – biphenyl]-3-carboxamide

Compound 44
[Step 1 : Synthesis of 5-brom -2-methyl-3-nitrobenzoic acid

To stirred solution of 2-methyl-3-nitrobenzoic acid ( 100 g, 552 mmol) in cone. H2S04 (400 mL), 1 ,3-dibromo-5,5-dimethyl-2,4-imidazolidinedione (88 g, 308 mmol) was added in a portion wise manner at room temperature and the reaction mixture was then stirred at room temperature for 5 h. The reaction mixture was poured onto ice cold water, the precipitated solid was filtered off, washed with water and dried under vacuum to afford the desired compound as a solid ( 140 g, 98%). The isolated compound was taken directly into the next step. Ή NMR (DMSO-4$, 400 MHz) δ 8.31 (s, 1 H), 8.17 (s, 1 H), 2.43 (s, 3H).
Step 2: Synthesis of methyl -bromo-2-methyl-3-nitrobenzoate

To a stirred solution of 5-bromo-2-methyl-3-nitrobenzoic acid (285 g, 1 105 mmol) in DMF (2.8L) at room temperature was added sodium carbonate (468 g, 4415 mmol) followed by addition of methyl iodide (626.6 g, 4415 mmol). The resulting reaction mixture was heated at 60 °C for 8 h. After completion (monitored by TLC), the reaction mixture was filtered (to remove sodium carbonate) and washed with ethyl acetate ( 1 L X 3). The combined filtrate was washed with water (3L X 5) and the aqueous phase was back extracted with ethyl acetate (1L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (290g, 97% yield). The isolated compound was taken directly into the next step. Ή NMR (CDC13, 400 MHz) δ 8.17 (s, 1H), 7.91 (s, 1H), 3.96 (s, 3H), 2.59 (s, 3H).
Step 3: Synthesis of methyl 3-amino-5-bromo-2-methylbenzoate

To a stirred solution of methyl 5-bromo-2-methyl-3-nitrobenzoate (290 g,
1058 mmol) in ethanol (1 .5L) was added aqueous ammonium chloride (283 g, 5290 mmol dissolved in 1.5L water). The resulting mixture was stirred at 80°C to which iron powder (472 g, 8451 mmol) was added in a portion wise manner. The resulting reaction mixture was heated at 80 °C for 12 h. Upon completion as determined by TLC, the reaction mixture was hot filtered over celite® and the celite bed was washed with methanol (5L) followed by washing with 30% MeOH in DCM (5L). The combined filtrate was concentrated in-vacuo, the residue obtained was diluted with aqueous sodium bicarbonate solution (2L) and extracted with ethyl acetate (5L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (220 g, 85%). The compound was taken directly into the next step. Ή NMR (CDC13, 400 MHz) δ 7.37 (s, 1 H), 6.92 (s, 1 H), 3.94 (s, 3H), 3.80 (bs, 2H), 2.31 (s, 3H).
Step 4: Synthesis of methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate

To a stirred solution of methyl 3-amino-5-bromo-2-methylbenzoate (15 g, 61 .5 mmol) and dihydro-2H-pyran-4(3)-one (9.2 g, 92 mmol) in dichloroethane (300 mL) was added acetic acid (22 g, 369 mmol) and the reaction mixture stirred at room temperature for 15 minutes, then the reaction mixture was cooled to 0°C and sodium triacetoxyborohydnde (39 g, 184 mmol) was added. The reaction mixture was stirred overnight at room temperature. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH of 7-8 was obtained. The organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100-200 mesh silica gel) eluting with ethyl acetate: hexane to afford the desired compound as a solid ( 14 g, 69%). ‘H NMR (DMSO-<fc, 400 MHz) δ 7.01 (s, 1 H), 6.98 (s, 1 H), 5.00 (d, 1 H, J=7.6 Hz), 3.84-3.87 (m, 2H), 3.79 (s, 31 1), 3.54-3.56 (mf 1 H), 3.43 (L 21 1, J 12 Hz), 2.14 (s. 31 1). 1 . 1 – 1 .84 (m: 211). 1 .47- 1 .55 (m, 2H).
Step 5: Synthesis of methyl 5-bromo-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2- methylbenzoate

To a stirred solution of methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate (14 g, 42.7 mmol) in dichloroethane (150 mL) was added acetaldehyde (3.75 g, 85.2 mmol) and acetic acid ( 15.3 g, 256 mmol). The resulting reaction mixture was stirred at room temperature for 15 minutes. The mixture was cooled to 0 °C and sodium
triacetoxyborohydnde (27 g, 128 mmol) was added. The reaction mixture was stirred at room temperature for 3 hours. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH 7-8 was obtained, the organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100- 200 mesh silica gel) eluting with ethyl acetate: hexane to afford the desired compound as a viscous liquid (14 g, 93%). Ή NMR (DMSO-cfo 400 MHz) δ 7.62 (s, 1 H), 7.52 (s, 1 H), 3.80 (bs, 5H), 3.31 (t, 2H), 2.97-3.05 (m, 2H), 2.87-2.96 (m, 1 H), 2.38 (s, 3H), 1.52-1.61 (m, 2H), 1 .37-1.50 (m, 2H), 0.87 (t, 3H, J=6.8 Hz).
Step 6: Synthesis of 5-bromo-N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-3 -(ethyl (tetrahydro-2H-pyra -4-yl) amino)-2-methylbenzamide

To a stirred solution of 5-bromo-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2- methylbenzoate (14 g, 39.4 mmol) in ethanol ( 100 mL) was added aqueous NaOH (2.36 g, 59.2 mmol in 25mL water) and the resulting mixture was stirred at 60 °C for 1 h. Upon completion of the reaction as determined by TLC, the solvent was removed under reduced pressure and the residue obtained was acidified with IN HC1 until a pH 7 was obtained and then aqueous citric acid solution was added until a pH 5-6 was obtained. The aqueous layer was extracted with 10% MeOH in DCM (200mL X 3), the combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give the respective acid (14 g, 100%).
The above acid (14 g, 40.9 mmol) was then dissolved in DMSO (70 mL) and 3- (amino methyl)-4, 6-dimethylpyridin-2( l H)-one ( 12.4 g, 81 .9 mmol) was added to it. The reaction mixture was stirred at room temperature for 15 minutes, then PYBOP (31.9 g, 61.4 mmol) was added and stirring was continued for overnight at room temperature. Upon completion of the reaction as determined by TLC, the reaction mixture was poured onto ice- cold water (700 mL), stirred for 30 minutes and the precipitated solid was collected by filtration, washed with water (500 mL) and air dried. The solid obtained was stirred with acetonitrile (75mL X 2), filtered and air dried. The solid obtained was again stirred with 5% MeOH in DCM ( l OOmL), filtered and dried completely under vacuum to afford the title compound as a solid ( 14 g, 74 %). Ή NMR (DMSO- 6, 400 MHz) δ 1 1.47 (s, 1 H), 8.23 (t, 1 H), 7.30 (s, 1 H), 7.08 (s, 1 H), 5.85 (s, 1 H), 4.23 (d, 2H, J=4.4 Hz), 3.81 (d, 2H, J=l 0.4 Hz), 3.20-3.26 (m, 2H), 3.00-3.07 (m, I H), 2.91 -2.96 (m, 2H), 2.18 (s, 3H), 2.14 (s, 3H), 2.10 (s, 3H), 1.58-1.60 (m, 2H), 1.45-1.50 (m, 2H), 0.78 (t, 3H, J=6.8 Hz).
Step 7: Synthesis of N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-5- (ethyl (tetrahydro-2H-pyran-4-yl) amino)-4-methyl-4′-(morpholinomethyl)-[l , l ‘-biphenyl]-3- carboxamide
 TITLE COMPD
TITLE COMPDTo a stirred solution of 5-bromo-N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2-methylbenzamide (14 g, 29.5 mmol) in dioxane/ water mixture (70 mL/ 14 mL) was added 4-(4-(4, 4, 5, 5-tetramethyl- l , 3, 2- dioxaborolan-2-yl) benzyl) morpholine (13.4 g, 44.2 mmol) followed by addition of Na2C03 (1 1 .2 g, 106.1 mmol). The solution was purged with argon for 15 minutes and then Pd (PPh3)4 (3.40 g, 2.94 mmol) was added and the solution was again purged with argon for a further 10 min. The reaction mixture was heated at 100°C for 4 h. After completion (monitored by TLC), the reaction mixture was diluted with water and extracted with 10% MeOH/DCM.
The combined organic layers were dried over anhydrous sodium sulphate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100- 200 mesh silica gel) eluting with methanol: DCM to the title compound as a solid (12 g, 71 %).
Analytical Data: LCMS: 573.35 (M + 1 )+; HPLC: 99.5% (@ 254 nm) (R,;3.999; Method: Column: YMC ODS-A 1 50 mm x 4.6 mm x 5 μ; Mobile Phase: A; 0.05% TFA in water/ B; 0.05% TFA in acetonitrile; Inj. Vol : 10 μΐ, Col. Temp.: 30 °C; Flow rate: 1 .4 mL/min.;
Gradient: 5% B to 95% B in 8 min, Hold for 1 .5 min, 9.51 -12 min 5% B);
Ή NMR (DMSO-i 6, 400 MHz) 5 1 1 .46 (s, I H), 8. 19 (t, 1 H), 7.57 (d, 2H, J=7.2 Hz), 7.36-7.39 (m, 3H), 7.21 (s, I H), 5.85 (s, I H), 4.28 (d, 2H, J=2.8 Hz), 3.82 (d, 2H, J=9.6 Hz), 3.57 (bs, 4H), 3.48 (s, 2H), 3.24 (t, 2H, J=10.8Hz), 3.07-3.09 (m, 2H), 3.01 (m, I H), 2.36 (m, 4H), 2.24 (s, 3H), 2.20 (s, 3H), 2.10 (s, 3H), 1 .64-1 .67 (m, 2H), 1 .51 – 1 .53 (m, 2H), 0.83 (t, 3H, J=6.4 Hz).
TRIHYDROCHLORIDE
Step 8: Synthesis of N-((4,6-dimethyl-2-oxo-l ,2-dihydropyridin-3-yl)methyl)-5- (ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[ 1 , 1 ‘-biphenyl]-3- carboxamide trihydrochloride

N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-5-(ethyl (tetrahydro- 21 l-pyran-4-yl) amino)-4-methyI-4′-(niorpholinornethyl)-[ 1 , 1 ‘-biphenyl]-3-carboxamide ( 12 g, 21.0 mmol) was dissolved in methanolic HC1 (200 mL) and stirred at room temperature for 3 h. After three hours of stirring, the reaction mixture was concentrated under reduced pressure. The solid obtained was stirred with ether ( l OOmL X 2) to afford the desired salt as a solid ( 1 1 g, 77 %).
Analytical Data of the tri-HCl salt: LCMS: 573.40 (M + 1 )+; HPLC: 99.1 % (@ 254 nm) (R,;3.961 ; Method: Column: YMC ODS-A 150 mm x 4.6 mm x 5 μ; Mobile Phase: A; 0.05% TFA in water/ B; 0.05% TFA in acetonitrile; Inj. Vol: 10 pL, Col. Temp.: 30 °C; Flow rate: 1.4 mL/min.; Gradient: 5% B to 95% B in 8 min, Hold for 1.5 min, 9.51 -12 min 5% B);
Ή NMR (D20 400 MHz) δ 7.92 (bs, I H,) 7.80 (s, I H), 7.77 (d, 2H, J=8 Hz), 7.63 (s, I H), 7.61 (s, I H), 6.30 (s, I H), 4.48 (s, 2H), 4.42 (s, 2H), 4.09-4.1 1 (m, 4H), 3.95-3.97 (m, 2H), 3.77 (t, 3H, J=10.4 Hz), 3.44-3.47 (m, 3H), 3.24-3.32 (m, 3H), 2.42 (s, 3H), 2.35 (s, 3H), 2.26 (s, 3H), 2.01 (m, 2H), 1 .76 (m, 2H), 1 .04 (t, 3H, J=6.8 Hz).
…………………………………………
PATENT
WO2013155317
http://www.google.com/patents/WO2013155317A1?cl=en
N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)-5-(ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’- biphenyl] -3-carboxamide hydrobromide:

N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-5-(ethyl
(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’-biphenyl]-3- carboxamide hydrobromide:

As used herein, “Compound I” refers to N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)-5-(ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’- biphenyl]-3-carboxamide. The hydrobromide of Compound I can be used to inhibit the histone methyltransferase activity of EZH2, either in a subject or in vitro. The hydrobromide of Compound I can also be used to treat cancer in a subject in need thereof.
Scheme 1

……………………………………..Compound I Compound I – HBr
HPLC
HPLC was conducted on an Agilent 1200 HPLC quaternary pump, low pressure mixing, with an in-line degasser. Analytical method conditions: 8 μΐ^ sample (20 mg of ER-581982-06 diluted with 50 mL of a methanol to provide approximately 0.4 mg/mL solution) was injected onto a Agilent Zorbax Eclipse XDB-C18 (4.6 x 150 mm, 3.5 um), Chromatography conditions: mobile phase A, water with 5mM ammonium formate; mobile phase B, 5 mM ammonium formate in 50/45/5 acetonitrile/methanol/water; flow rate, 1.5 ml/min.; gradient: isocratic at 10% B from 0 to 3 min; linear increase to 70% B from 3 to 7 min; isocratic at 70% B from 7 to 12 min; linear increase to 100% B from 12 to 15 min isocratic at 100% B from 15 to 20 min;
column temperature, 35 °C; detection, UV 230 nm. Approximate retention time of Compound I = 10.7 min.
Synthesis of Polymorph A

5-bromo-2-methyl-3-nitrobenzoic acid stirred solution of 2-methyl-3-nitrobenzoic acid (100 g, 552 mmol) in cone. H2S04 (400 mL), l,3-dibromo-5,5-dimethyl-2,4- imidazolidinedione (88 g, 308 mmol) was added in a portion wise manner at room temperature and the reaction mixture was then stirred at room temperature for 5 h. The reaction mixture was poured onto ice cold water, the precipitated solid was filtered off, washed with water and dried under vacuum to afford the desired compound as a solid (140 g, 98%). The isolated compound was taken directly into the next step. 1H NMR (DMSO-J6, 400 MHz) δ 8.31 (s, 1H), 8.17 (s, 1H), 2.43 (s, 3H).

Methyl 5-bromo-2-methyl-3-nitrobenzoate To a stirred solution of 5-bromo-2- methyl-3-nitrobenzoic acid (285 g, 1105 mmol) in DMF (2.8L) at room temperature was added sodium carbonate (468 g, 4415 mmol) followed by addition of methyl iodide (626.6 g, 4415 mmol). The resulting reaction mixture was heated at 60 °C for 8 h. After completion (monitored by TLC), the reaction mixture was filtered (to remove sodium carbonate) and washed with ethyl acetate (1L X 3). The combined filtrate was washed with water (3L X 5) and the aqueous phase was back extracted with ethyl acetate (1L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (290g, 97% yield). The isolated compound was taken directly into the next step. 1H NMR (CDC13, 400 MHz) δ 8.17 (s, 1H), 7.91 (s, 1H), 3.96 (s, 3H), 2.59 (s, 3H).

Methyl 3-amino-5-bromo-2-methylbenzoate (1) To a stirred solution of methyl 5- bromo-2-methyl-3-nitrobenzoate (290 g, 1058 mmol) in ethanol (1.5L) was added aqueous ammonium chloride (283 g, 5290 mmol dissolved in 1.5L water). The resulting mixture was stirred at 80°C to which iron powder (472 g, 8451 mmol) was added in a portion wise manner. The resulting reaction mixture was heated at 80 °C for 12 h. Upon completion as determined by TLC, the reaction mixture was hot filtered over celite® and the celite bed was washed with methanol (5L) followed by washing with 30% MeOH in DCM (5L). The combined filtrate was concentrated in- vacuo, the residue obtained was diluted with aqueous sodium bicarbonate solution (2L) and extracted with ethyl acetate (5L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (220 g, 85%). The compound was taken directly into the next step. 1H
NMR (CDCI3, 400 MHz) δ 7.37 (s, 1H), 6.92 (s, 1H), 3.94 (s, 3H), 3.80 (bs, 2H), 2.31 (s, 3H).

Methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate (2) A reactor was charged with methyl 3-amino-5-bromo-2-methylbenzoate (455.8 g, 1.87 mol), 1,2- Dichloroethane (4.56 L), and acetic acid (535 ml, 9.34 mol). To the mixture were added dihydro-2H-pyran-4(3H)-one (280 g, 2.80 mol) and sodium triacetoxyborohydride (594 g, 2.80 mol) maintaining the internal temperature below 40 °C. The mixture was stirred at 25 °C for 2.5 h and then the reaction was quenched with a solution of sodium hydroxide (448 g, 11.20 mol) in water (5.61 L). After stirring for 20 minutes at ambient temperature, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (3.65 L). The organic layers were combined, washed with brine (1.5 L), and concentrated under vacuum.
The residue was treated with ethyl acetate (1.8 L) and heated to 65-70 °C. The mixture was stirred at 65-70 °C for 15 minutes to give a clear solution and then treated with n-heptane (7.3 L) maintaining the temperature between 60-70 °C. Once the heptane was completely added to the solution, the mixture was held at 65-70 °C for 15 minutes and then allowed to cool to 18- 22 °C over 3 h. The resulting suspension was stirred at 18-22 °C for 4 h, cooled to 0-5 °C over 1 h, and held at 0-5 °C for 2 h. The precipitate was filtered, washed twice with n-heptane (1.4 L), and dried under vacuum to give the title compound (540 g, 88%). The XRPD pattern of this compound is shown in Figure 17.

Methyl 5-bromo-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2-methylbenzoate (3)
To a stirred solution of methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate (14 g, 42.7 mmol) in dichloroethane (150 mL) was added acetaldehyde (3.75 g, 85.2 mmol) and acetic acid (15.3 g, 256 mmol). The resulting reaction mixture was stirred at room temperature for 15 minutes. The mixture was cooled to 0 °C and sodium triacetoxyborohydride (27 g, 128 mmol) was added. The reaction mixture was stirred at room temperature for 3 hours. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH 7-8 was obtained, the organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100-200 mesh silica gel) eluting with ethyl acetate: hexane to afford the desired compound as a viscous liquid (14 g, 93%). 1H NMR DMSO-d6, 400 MHz) δ 7.62 (s, 1H), 7.52 (s, 1H), 3.80 (bs, 5H), 3.31 (t, 2H), 2.97-3.05 (m, 2H), 2.87-2.96 (m, 1H), 2.38 (s, 3H), 1.52-1.61 (m, 2H), 1.37-1.50 (m, 2H), 0.87 (t, 3H, J=6.8 Hz).

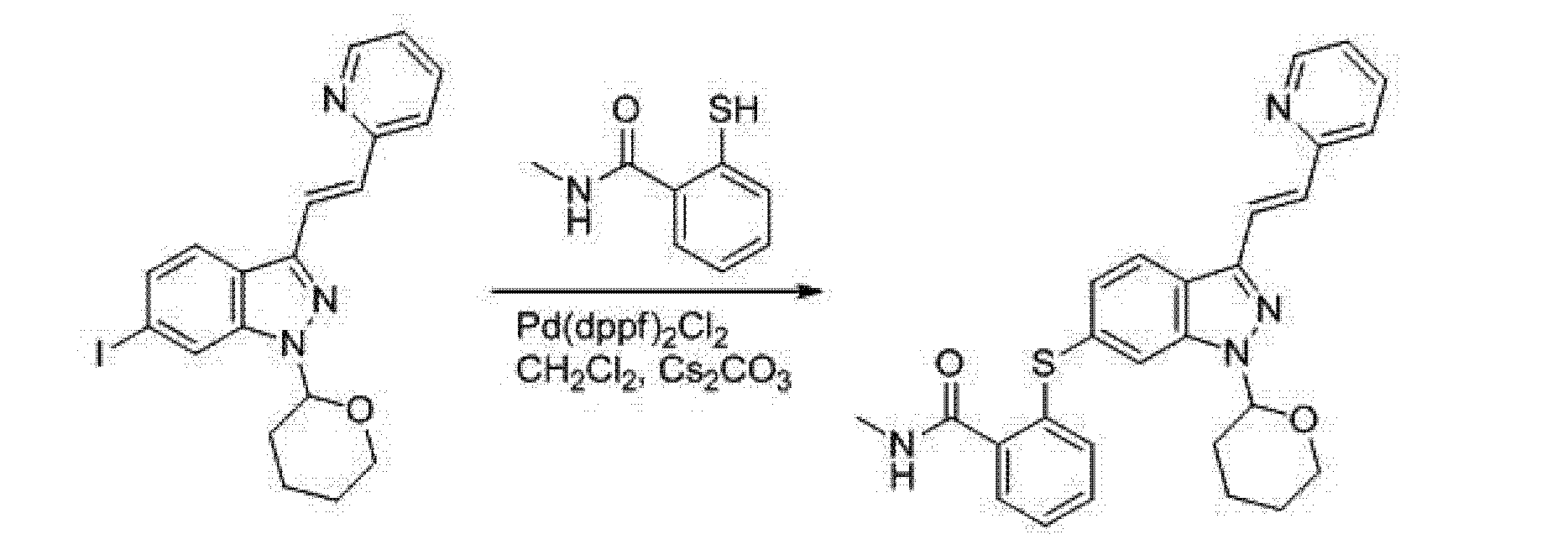
Methyl 5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-
[l,l’-biphenyl]-3-carboxylate (4): A mixture of methyl 5-bromo-3-(ethyl(tetrahydro-2H-pyran- 4-yl)amino)-2-methylbenzoate (580 g, 1.63 mol), 4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan- 2-yl)benzyl)morpholine (592 g, 1.95 mol), 1,4-dioxane (3.86 L), sodium carbonate (618 g, 5.83 mol), and water (771 ml) was degassed by bubbling nitrogen through the mixture at 20 °C for 20 minutes and treated with tetrakis(triphenylphosphine)palladium(0) (14.11 g, 12.21 mmol). The resulting mixture was degassed for an additional 20 minutes and then heated to 87-89 °C for 17 h. After cooling to 20 °C, the mixture was diluted with ethyl acetate (5.80 L) and a solution of (R)-2-Amino-3-mercaptopropionic acid (232 g) in water (2.320 L). After stirring for 1 h at 20 °C, the organic layer was separated and washed again with a solution of (R)-2-Amino-3- mercaptopropionic acid (232 g) in water (2.320 L). The aqueous layers were combined and extracted with ethyl acetate (5.80 L). The organic layers were combined, washed with a solution of sodium hydroxide (93 g) in water (2.32 L), and concentrated under vacuum at 35 °C to give the title compound as an orange oil (1.21 kg, 164% yield).

5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’- biphenyl]-3-carboxylic acid (5): Methyl 5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′- (morpholinomethyl)-[l,l’-biphenyl]-3-carboxylate (69.0 g, 152.5 mmol) (based on the theoretical yield from the previous step) was suspended in ethanol (380 mL) and treated with a solution of sodium hydroxide (24.84 g, 621.0 mmol) in water (207 mL). The mixture was stirred at 40°C for 18 h. After cooling to 0-5 °C, the mixture was neutralized to pH 6.5 with 1 N hydrochloric acid (580 mL) maintaining the temperature below 25 °C. Then, the mixture was extracted twice with a mixture of dichloromethane (690 mL) and methanol (69.0 mL). The organic layers were combined and concentrated under vacuum to give a crude product as a yellow solid (127g).
The crude product was dissolved in 2-methyltetrahydrofuran (656 mL) at 70 °C and then treated with IPA (828 mL). The mixture was allowed to cool to rt over 3-4 h and then stirred overnight at rt. The precipitate was filtered, washed twice with IPA (207 mL), and dried under vacuum to give the title compound as an off white solid (53.54 g, 80%). The XRPD pattern of this compound is shown in Figure 9.

N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-5-(ethyl(tetrahydro-2H- pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’-biphenyl]-3-carboxamide
(Compound I): A mixture of 5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′- (morpholinomethyl)-[l,l’-biphenyl]-3-carboxylic acid (540 g, 1.23 mol) and 3-(aminomethyl)- 4,6-dimethyl-dihydro-pyridin-2(lH)-one hydrochloride (279 g, 1.48 mol) was suspended in DMSO (2.70 L) and treated with triethylamine (223 ml, 1.60 mol). The mixture was stirred at 25 °C for 30 min and treated with EDC-HC1 (354 g, 1.85 mol) and HOBT hydrate (283 g, 1.85 mol). The reaction mixture was stirred at rt for 16 h. After addition of triethylamine (292 ml, 2.09 mol), the mixture was cooled to 15 °C, diluted with water (10.1 L) maintaining the temperature below 30 °C, and stirred at 19-25 °C for 4 h. The resulting precipitate was filtered, washed twice with water (2.70 L), and dried under vacuum to give a crude product (695 g, wt-wt analysis = 78%).
For the further purification of the product, recrystallization was conducted. A crude product (20.00 g, 34.92 mmol) was suspended in a mixture of ethanol (190 ml) and water (10.00 ml) and heated to 75°C until a clear solution was obtained. The solution was allowed to cool to rt overnight. The precipitate was filtered, washed twice with a mixture of ethanol (30.0 ml) and water (30.0 ml), and dried under vacuum at 35 °C to give the title compound as an off white solid (14.0 g, 70% recovery from the crude and 90% yield based on wt-wt assay).

4-((3′-(((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′- (ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[l,l’-biphenyl]-4-yl)methyl)morpholin- 4-ium bromide (Polymorph A): A crude N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)am
biphenyl]-3-carboxamide (595 g, 464 g based on wt-wt assay, 810.3 mmol) was suspended in ethanol (3.33 L). After heating to 70 °C, the mixture was treated with 48% aqueous HBr (97 ml, 850.8 mmol) and stirred at 70 °C for 30 min. The resulting orange-red solution was treated with ethyl acetate (3.33 L) maintaining the temperature above 60 °C. The mixture was slowly cooled to rt over 18 h. The mixture was cooled to 0 °C over 1 h and stirred at that temperature for 5.5 h. The resulting precipitate was filtered, washed twice with ethyl acetate (1.39 L), and dried under vacuum to give the title compound as an off white solid (515 g, 97% yield).
Recrystallization of Polymorph A: 4-((3′-(((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[l,l’-biphenyl]-4- yl)methyl)morpholin-4-ium bromide (0.50 g, 0.77 mmol; 95.6% pure by HPLC) was suspended in ethanol (3.0 mL) and heated to 80 °C until a clear solution was obtained. To the solution was added MTBE (5.0 mL) slowly. The resulting solution was allowed to cool to 18-22 °C over 3 h and stirred at 18-22 °C for 15 h. The precipitate was filtered, washed twice with MTBE (2 mL) and dried under vacuum to give 0.45 g of the title compound (89% recovery, 96.6% pure by HPLC).
Compound I is protonated at the nitrogen of the morpholino substituent, providing a monohydrobromide of Compound I having the following structure:

This particular monohydrobromide can be referred to as “4-((3′-(((4,6-dimethyl-2-oxo- l,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′- methyl-[l, -biphenyl]-4-yl)methyl)morpholin-4-ium bromide.” Figure 11 depicts the X-ray crystal structure of this particular salt form.
…………………………………………………………..
see
WO-2015057859
Eisai Research Institute; Epizyme Inc
Novel crystalline polymorphic form C of tazemetostat, useful for treating an EZH2-mediated cancer, including non-Hodgkin’s lymphoma and breast cancer.
…………………
PAPER
RSC Advances (2015), 5(33), 25967-25978
http://pubs.rsc.org/en/content/articlelanding/2015/ra/c5ra02365c#!divAbstract
DOI: 10.1039/C5RA02365C
The histone lysine methyltransferase EZH2 has been implicated as a key component in cancer aggressiveness, metastasis and poor prognosis. This study discovered a new class of hexahydroisoquinolin derivatives as EZH2 inhibitors. A structure–activity relationship study showed that the steric hindrance was important to the activity for EZH2. A preliminary optimization study led to the discovery of several potent compounds with low nanomolar to sub-nanomolar potency for EZH2. Biological evaluation indicated that SKLB1049 was a highly potent with improved solubility compared to EPZ6438, SAM-competitive, and cell-active EZH2 inhibitor that decreased global H3K27me3 in SU-DHL-6 and Pfeiffer lymphoma cells in a concentration- and time-dependent manner. Further study indicated that SKLB1049 caused cell arrest in G0/G1 phase. These compounds would be useful as chemical tools to further explore the biology of EZH2 and provided us with a start point to develop new EZH2 inhibitors.

|
In vitro protocol: |
Proc Natl Acad Sci U S A. 2013 May 7;110(19):7922-7. |
|
In vivo protocol: |
Proc Natl Acad Sci U S A. 2013 May 7;110(19):7922-7. |
|
References |
1: Knutson SK, Warholic NM, Johnston LD, Klaus CR, Wigle TJ, Iwanowicz D, Littlefield BA, Porter-Scott M, Smith JJ, Moyer MP, Copeland RA, Pollock RM, Kuntz KW, Raimondi A, Keilhack H. Synergistic Anti-Tumor Activity of EZH2 Inhibitors and Glucocorticoid Receptor Agonists in Models of Germinal Center Non-Hodgkin Lymphomas. PLoS One. 2014 Dec 10;9(12):e111840. doi: 10.1371/journal.pone.0111840. eCollection 2014. PubMed PMID: 25493630; PubMed Central PMCID: PMC4262195.
2: Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Waters NJ, Smith JJ, Porter-Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Uenaka T, Pollock RM, Kuntz KW, Yokoi A, Keilhack H. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014 Apr;13(4):842-54. doi: 10.1158/1535-7163.MCT-13-0773. Epub 2014 Feb 21. PubMed PMID: 24563539
3. Inhibitors of human histone methyltransferase EZH2, and methods of use thereof for treating cancer. By Kuntz, Kevin W.; Knutson, Sarah K.; Wigle, Timothy James Nelson . From U.S. Pat. Appl. Publ. (2013), US 20130040906 A1 20130214.
4. Aryl-or heteroaryl-substituted benzamide compounds as anticancer agents and their preparation By Kuntz, Kevin Wayne; Chesworth, Richard; Duncan, Kenneth William; Keilhack, Heike; Warholic, Natalie; Klaus, Christine; Zheng, Wanjun; Seki, Masashi; Shirotori, Syuji; Kawano, Satoshi From PCT Int. Appl. (2012), WO 2012142504 A1 20121018.
5: Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM, Kuntz KW, Keilhack H. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013 May 7;110(19):7922-7. doi: 10.1073/pnas.1303800110. Epub 2013 Apr 25. PubMed PMID: 23620515; PubMed Central PMCID: PMC3651445.
| WO2013155317A1 * | Apr 11, 2013 | Oct 17, 2013 | Epizyme, Inc. | Salt form of a human hi stone methyltransf erase ezh2 inhibitor |
| WO2013155464A1 * | Apr 12, 2013 | Oct 17, 2013 | Epizyme, Inc. | Combination therapy for treating cancer |
| WO2014049488A1 * | Sep 16, 2013 | Apr 3, 2014 | Pfizer Inc. | Benzamide and heterobenzamide compounds |
| WO2014062732A1 * | Oct 15, 2013 | Apr 24, 2014 | Epizyme, Inc. | Substituted benzene compounds |
| WO2014062733A2 * | Oct 15, 2013 | Apr 24, 2014 | Epizyme, Inc. | Substituted benzene compounds |
| WO2014172044A1 * | Mar 14, 2014 | Oct 23, 2014 | Epizyme, Inc. | Substituted benzene compounds |
| WO2015004618A1 * | Jul 9, 2014 | Jan 15, 2015 | Glaxosmithkline Intellectual Property (No.2) Limited | Enhancer of zeste homolog 2 inhibitors |
| WO2015010049A1 * | Jul 18, 2014 | Jan 22, 2015 | Epizyme, Inc. | Substituted benzene compounds |
| WO2015010078A2 | Jul 18, 2014 | Jan 22, 2015 | Epizyme, Inc. | Substituted 6,5-fused bicyclic heteroaryl compounds |
| WO2011140325A1 * | May 5, 2011 | Nov 10, 2011 | Glaxosmithkline Llc | Indazoles |
| WO2012142504A1 * | Apr 13, 2012 | Oct 18, 2012 | Eisai Co., Ltd. | Aryl-or heteroaryl-substituted benzene compounds |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014062720A2 * | Oct 15, 2013 | Apr 24, 2014 | Epizyme, Inc. | Methods of treating cancer |
| WO2011140324A1 * | May 5, 2011 | Nov 10, 2011 | Glaxosmithkline Llc | Indoles |
| WO2011140325A1 * | May 5, 2011 | Nov 10, 2011 | Glaxosmithkline Llc | Indazoles |
| WO2012005805A1 * | May 5, 2011 | Jan 12, 2012 | Glaxosmithkline Llc | Azaindazoles |
| US4522811 | Jul 8, 1982 | Jun 11, 1985 | Syntex (U.S.A.) Inc. | Serial injection of muramyldipeptides and liposomes enhances the anti-infective activity of muramyldipeptides |
| US5763263 | Jul 24, 1996 | Jun 9, 1998 | Dehlinger; Peter J. | Method and apparatus for producing position addressable combinatorial libraries |
| US7563589 | May 27, 2005 | Jul 21, 2009 | The University Of North Carolina At Chapel Hill | Including EED, EZH2 and SUZ12 wherein the reconstituted complex has histone methyltransferase (HMTase) activity for lysine 27 of histone H3 (H3-K27); cancer |
Epizyme®, Inc.
400 Technology Square, 4th Floor
Cambridge, MA 02139
Phone: (617) 229-5872
Fax: (617) 349-0707
contact@Epizyme.com
![]()


 Jason Rhodes (left) has been appointed to president of Epizyme Inc.,
Jason Rhodes (left) has been appointed to president of Epizyme Inc.,

100 Technology Square

Filed under: cancer, PHASE1, Uncategorized Tagged: CANCER, eisai, epizyme, EPZ 6438, PHASE 1, tazemetostat






















 .
.
 .
.










































































































































.gif)




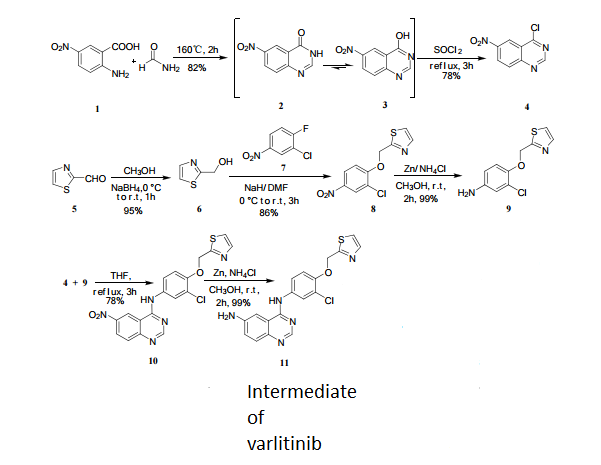
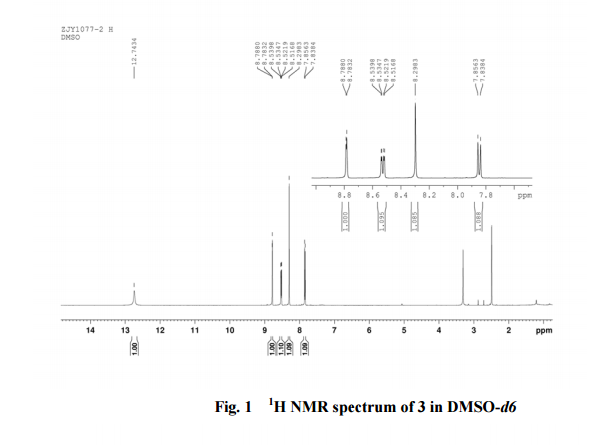
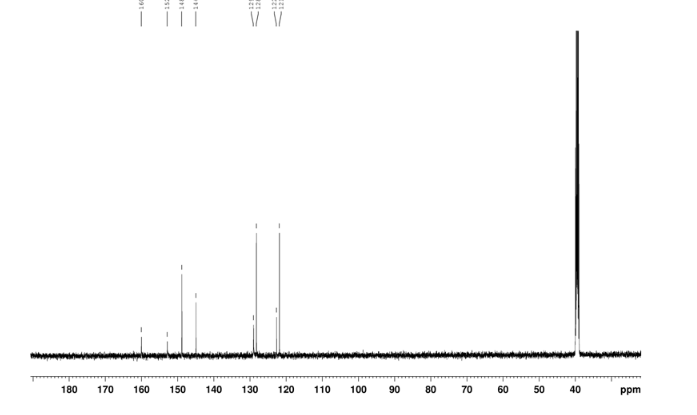
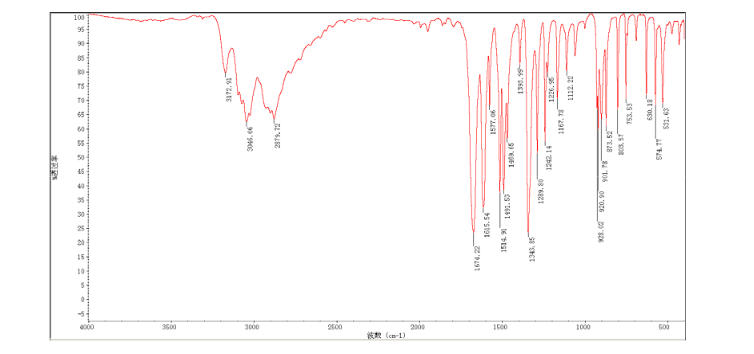
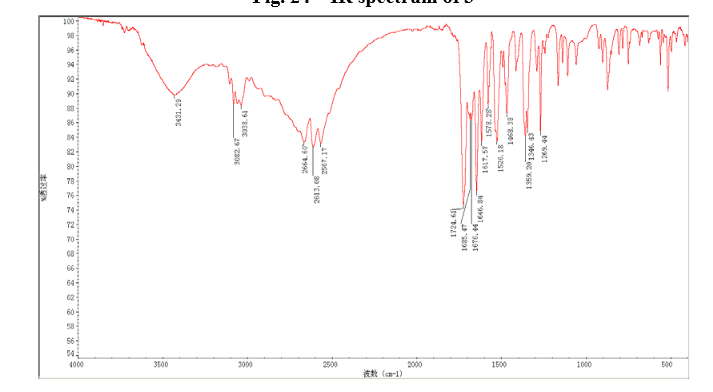
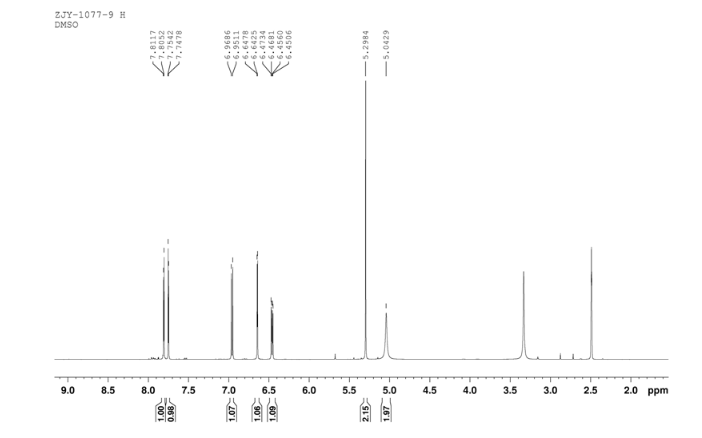
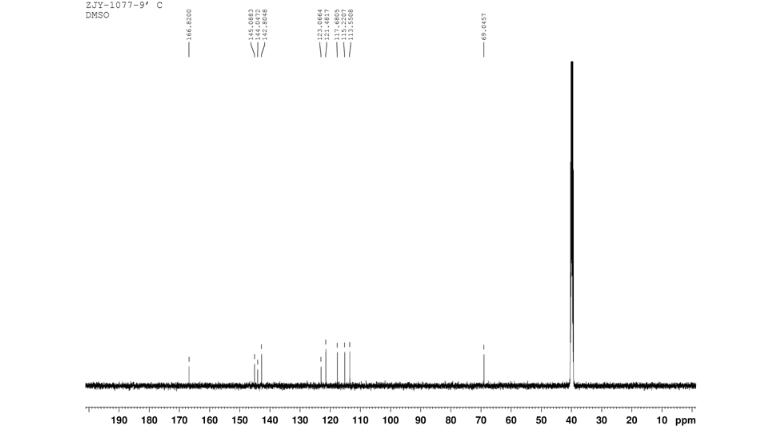
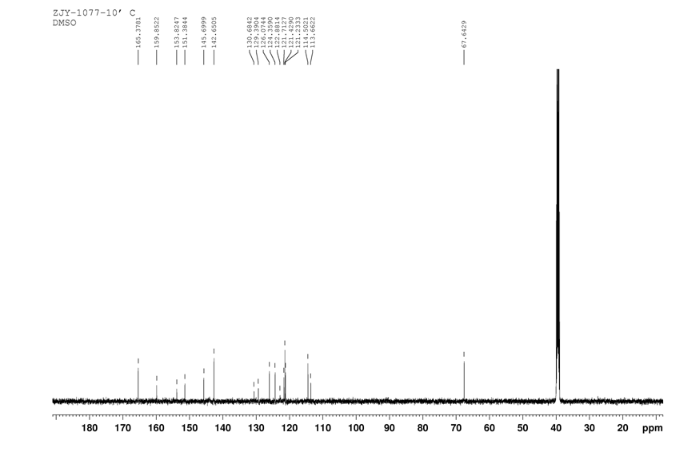
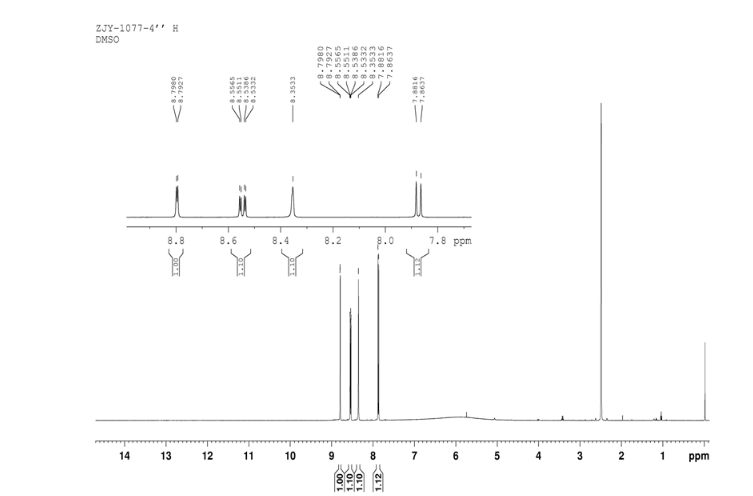 4 nmr dmsod6
4 nmr dmsod6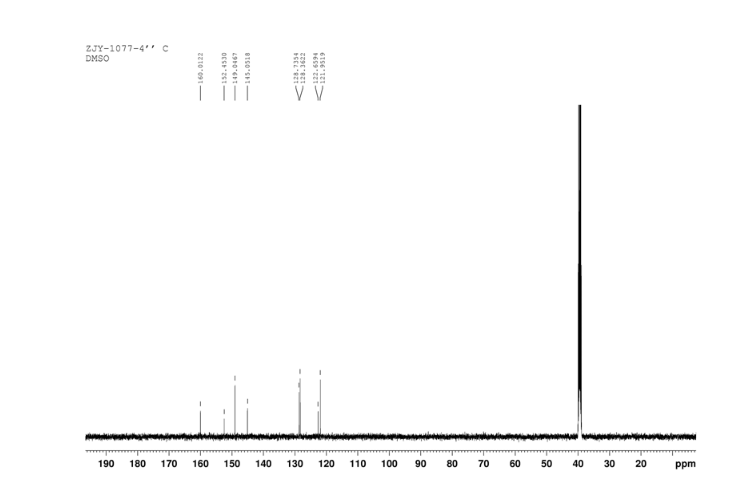
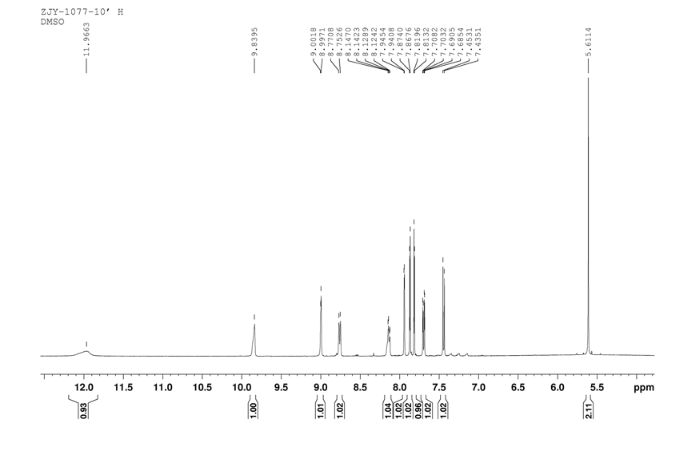
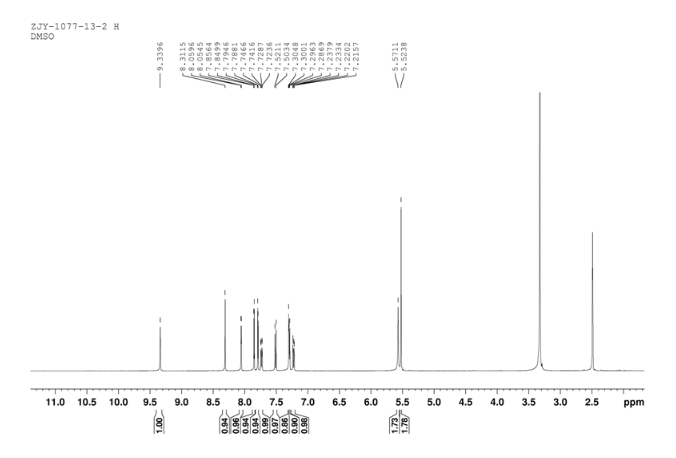
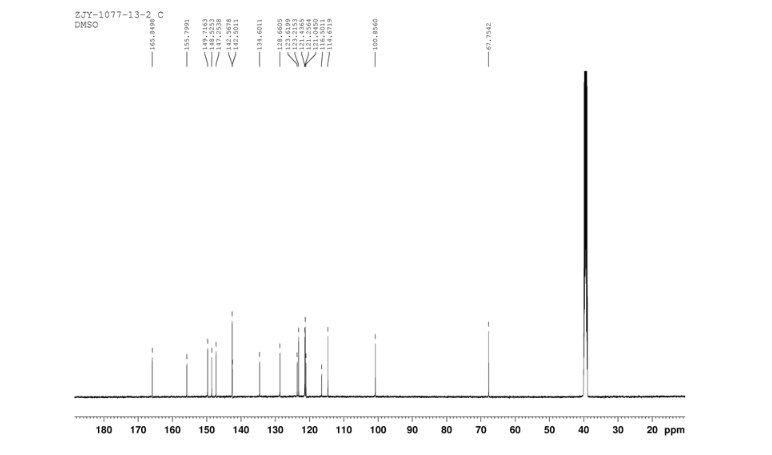
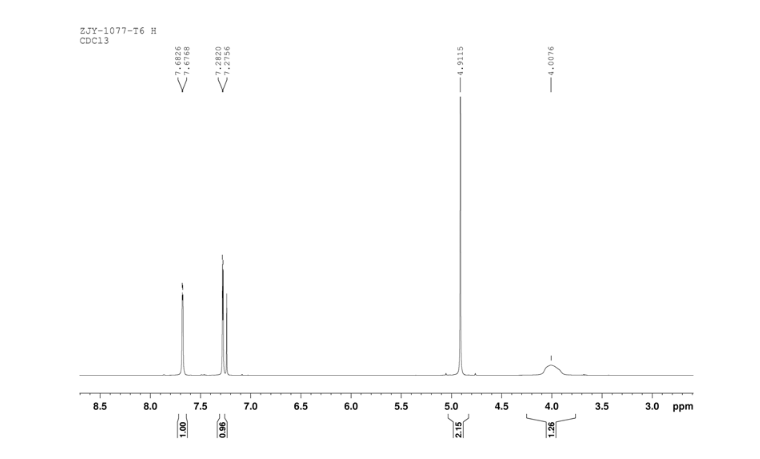 6 in dmsod6 1H NMR
6 in dmsod6 1H NMR
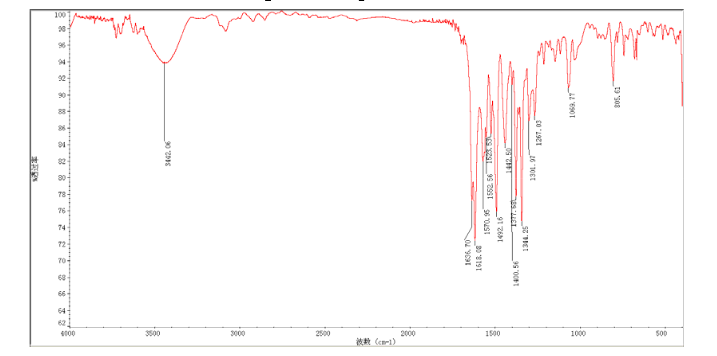


 carl fith
carl fith



























































%20structure.gif)





















![[1860-5397-11-135-1]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-11-135-1.png?max-width=550&background=EEEEEE)

![[1860-5397-11-135-i1]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-135-i1.png?max-width=550&background=EEEEEE)
![[1860-5397-11-135-i2]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-135-i2.png?max-width=550&background=EEEEEE)
![[1860-5397-11-135-i3]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-135-i3.png?max-width=550&background=EEEEEE)
![[1860-5397-11-135-i4]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-11-135-i4.png?max-width=550&background=EEEEEE)







































































 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..



 amcrasto@gmail.com
amcrasto@gmail.com


































































.png)

























































