CAS 1776112-90-3
Chemical Formula: C18H22FN9O2
Exact Mass: 415.188
Recruiting, Phase I/II (NTC02349633)
Epidermal growth factor receptor antagonists
Antineoplastics
Non-small cell lung cancer
Dose escalation study to evaluate safety, PK, PD and efficacy in advanced EGFRm+ NSCLC
02 May 2015Phase-I clinical trials in Non-small cell lung cancer (Metastatic disease, Second-line therapy or greater) in USA (PO) (NCT02349633)
05 Feb 2015Pfizer plans a phase I trial for Non-small cell lung cancer (Second-line therapy or greater) in USA (NCT02349633)
05 Jan 2015Preclinical trials in Non-small cell lung cancer in USA (PO)
PF-06747775 is an orally available inhibitor of the epidermal growth factor receptor (EGFR) mutant form T790M, with potential antineoplastic activity. EGFR T790M inhibitor PF-06747775 specifically binds to and inhibits EGFR T790M, a secondarily acquired resistance mutation, which prevents EGFR-mediated signaling and leads to cell death in EGFR T790M-expressing tumor cells. Compared to some other EGFR inhibitors, PF-06747775 may have therapeutic benefits in tumors with T790M-mediated drug resistance.
for the oral treatment of patients with locally advanced or metastatic EGFR mutant (del19 or L858R) non-small cell lung cancer
Kinetic mechanism for two-step covalent inhibition of EGFR
A suspension of 6-chloro-2-fluoro-9H-purine (5.49 g, 31.8 mmol, 1.00 eq), 3-methoxy-1-methyl-1H-pyrazol-4-amine hydrochloride (6.60 g, 40.34 mmol, 1.26 eq), and N,N-diisopropylethylamine (16.6 mL, 95.5 mmol, 3.00 eq) in DMSO (31.8 mL) was stirred at ambient temperature for 19 hr. The reaction mixture was then concentrated in vacuo at 50° C., poured into water (250 mL), and stirred vigorously at 0° C. for 1 hr. The resulting solids were filtered off, washed with ice cold water (20 mL), and dried for 16 hr at 50° C. to give the title compound (7.26 g, 87% yield, 96% purity) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 13.03 (br. s., 1 H) 9.21 (br. s., 1 H) 8.18 (br. s., 1 H) 7.74 (br. s., 1 H) 3.81 (br. s., 3 H) 3.71 (s, 3H). m/z (APCI+) for C10H11FN7O 264.2 (M+H)+.
Step 2: Preparation of 2-fluoro-N-(3-methoxy-1-methyl-1H-pyrazol-4-yl)-9-methyl -9H-purin-6-amine
To a vigorously stirred suspension of 2-fluoro-N-(3-methoxy-1-methyl-1H-pyrazol-4-yl)-9H-purin-6-amine (7.25 g, 27.5 mmol, 1.00 eq) and potassium carbonate (7.61 g, 55.1 mmol, 2.00 eq) in 1,4-dioxane (92.0 mL), was added dimethyl sulfate (2.90 mL, 30.3 mmol, 1.10 eq) in a dropwise manner over 3 min. After 4 hr, additional portions of 1,4-dioxane (50.0 mL), potassium carbonate (3.80 g, 27.5 mmol, 1.00 eq), and dimethyl sulfate (1.00 mL, 10.4 mmol, 0.30 eq) were added to the reaction mixture. After a further 16 hr, the reaction mixture was concentrated in vacuo, diluted with water (120 mL), and stirred at ambient temperature for 1 hr. The resulting solids were filtered, washed with water (20 mL), and dried for 16 hr at 60° C. to give the title compound (6.42 g, 84% yield, >95% purity) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.23 (br. s., 1 H) 8.13 (br. s., 1 H) 7.67 (s, 1 H) 3.78 (s, 3 H) 3.70 (s, 3 H) 3.69 (br. s., 3 H). m/z (APCI+) for C11H13FN7O 278.2 (M+H)+.
Step 3: Preparation of N-((3R,4R)-4-fluoro-1-(6-((3-methoxy-1-methyl-1H-pyrazol -4-yl)amino)-9-methyl-9H-purin-2-yl)pyrrolidin-3-yl)acrylamide
To a stirred suspension of 2-fluoro-N-(3-methoxy-1-methyl-1H-pyrazol-4-yl)-9-methyl-9H-purin-6-amine (554 mg, 2.00 mmol, 1.00 eq) and N-((3R,4R)-4-fluoropyrrolidin-3-yl)-3-(methylsulfonyl)propanamide (500 mg, 2.10 mmol, 1.05 eq) in DMSO (4.2 mL) was added N,N-diisopropylethylamine (0.83 mL, 5.00 mmol, 2.50 eq). The reaction mixture was then heated at 100° C. for 16 hr, cooled to ambient temperature, diluted with THF (4 mL), and treated with potassium tert-butoxide (4.00 mL, 1 M in THF, 2.00 eq). After 1 hr, an additional portion of potassium tert-butoxide (0.50 mL, 1 M in THF, 0.25 eq) was added to the reaction mixture. After a further 1 hr, the reaction mixture was poured into phosphate buffer (50 mL, pH=7) and water (50 mL), and extracted with ethyl acetate (5×40 mL). The combined organic layers were combined, dried (Na2SO4), and concentrated under reduced pressure. This crude product was then dissolved in ethyl acetate (40 mL) at 60° C. and then treated with heptanes (20 mL), at which point the solution became cloudy and was allowed to cool to ambient temperature and then to 0° C. After 16 hr at 0° C., the resulting solids were filtered and dried at ambient temperature to give the title compound (620.5 mg, 75% yield) as a white powder. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.44 (d, J=6.5 Hz, 1 H) 7.97 (s, 1 H) 7.82 (s, 1 H) 7.78 (s, 1 H) 6.23 (dd, J=10.0, 17.0 Hz, 1 H) 6.14 (dd, J=2.8, 17.0 Hz, 1 H) 5.62 (dd, J=2.8, 10.0 Hz, 1 H) 5.12 (d, J=51.0 Hz, 1 H) 4.46 (td, J=6.0, 11.9 Hz, 1 H) 3.88-3.6 (m, 4 H) 3.82 (s, 3 H) 3.71 (s, 3 H) 3.62 (s, 3 H). m/z (APCI+) for C18H23FN9O2 416.3 (M+H)+.
Example 7A
(Scheme F): Preparation of N-((3R,4R)-4-fluoro-1-(6-((3-methoxy-1-methyl-1H-pyrazol-4-yl)amino)-9-methyl-9H-purin-2-yl)pyrrolidin-3-yl)acrylamide
A mixture of xylene, (1.2 L), benzylamine (120 g, 1.10 mol, 1.0 eq) and L-(+)-tartaric acid (173 g, 1.15 mol, 1.05 eq) were heated at 135° C. for 12 hr (flask jacket temperature). Upon reaction completion, the mixture was cooled to 65° C. and MeOH (120 mL, 1 vol) was added. The resulting mixture was stirred for 1 hr and the resulting suspension was cooled to 20° C. followed by the addition of EtOAc (480 mL). Stirring was continued at 10° C. for 2 hr. The crude product was isolated by filtration and washed with EtOAc (120 mL) and dried on the filter. The crude product was then taken up in MeOH (480 mL) and heated at a gentle reflux for 1 hr, then cooled to 20° C. and granulated for 1 hr. The suspension was filtered and the precipitate washed with MeOH (240 mL) and dried to give the title compound (191 g, 864 mmol, 79%) as a white granular solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.38-7.30 (m, 2H) 7.30-7.22 (m, 3 H) 6.32 (br. s., 1 H) 4.59 (d, J=14.8 Hz, 1 H) 4.53 (d, J=14.8 Hz, 1 H) 4.40 (br. D., J=4.3 Hz, 2 H). m/z (EI+) for C11H11NO4 221.0 (M)+.
Preparation Step 2A: Preparation of (3S,4S)-1-benzylpyrrolidine-3,4-diol
To a mixture of (3R,4R)-1-benzyl-3,4-dihydroxypyrrolidine-2,5-dione (44 g, 199 mmol, 1.0 eq) and THF (176 mL) at 20° C. (vessel jacket temperature) was added borane-tetrahydrofuran complex (1.0 mol/L) in THF (800 mL, 800 mmol, 1.0 mol/L, 4.0 eq) at a rate to maintain the temperature between 20° C. and 25° C. Over 1 hr, the jacket temperature was ramped to 60° C. and then held for 1 hr. Upon completion, the reaction was cooled to 30° C. and quenched by the slow dropwise addition of MeOH (97 mL, 12 eq) to the mixture at a rate to control off gassing. The reaction mixture was then heated to reflux and concentrated to a low stir volume. The reaction solvent THF was then replaced by a constant volume displacement with MeOH (total of 1.5 L). Once the THF content had been reduced to less than 1 wt %, MeOH was replaced by a constant volume displacement with EtOAc (total of 1.5 L) to reduce the MeOH content to less than 1 wt %. The total volume of EtOAc was then readjusted to about 250 mL (6 vol) and then cooled to 5° C. to crystallize the product. The desired product was isolated by filtration, washed with cold EtOAc (88 mL) and dried to give title compound (27.0 g, 140 mmol, 70%). A second crop of product was isolated by concentration of the combined filtrate and cake wash to half volume, which was then cooled to 5° C., filtered and washed with cold EtOAc (50 mL) to afford additional title compound (4.5 g, 23 mmol, 12%). 1H NMR (400 MHz, DMSO-d6) δ ppm 7.33-7.26 (m, 4 H) 7.25-7.20 (m, 1 H) 4.48 (d, J=4.8 Hz, 2 H) 3.38-3.31 (m, 2 H), 3.57 (d, J=13.0 Hz, 1 H) 3.46 (d, J=13.0 Hz, 1 H) 2.74 (dd, J=9.4, 5.9 Hz, 2 H) 2.30 (dd, J=9.4, 4.4 Hz, 2 H). m/z (EI+) for C11H15NO2 194.2 (M+H)+.
Preparation Step 3A: Preparation of (3aR,6aS)-5-benzyl-2,2-dioxo-tetrahydro-1-oxa-2λ6-thia-3-5-diaza-pentalene-3-carboxylic acid t-butyl ester
To a 5 L jacketed reactor (Reactor 1) was added 1,4-dioxane (1.8 L), (3S,4S)-1-benzylpyrrolidine-3,4-diol (180 g, 0.932 mol, 1.0 eq) and TEA (792 mL, 5.68 mol, 6.1 eq) and the resulting mixture stirred at 10° C.
To a 2 L jacketed reactor (Reactor 2) was added 1,4-dioxane (1.6 L) and chlorosulfonyl isocyanate (596 g, 2.80 mol, 3.0 eq) and the resulting solution was cooled to 10° C. A solution of tert-butanol (211 g, 2.85 mol, 3.05 eq) in 1,4-dioxane (180 mL) was added over 45 min while maintaining the temperature between 10° C. and 20° C., and the resulting solution was then stirred for 15 min at 10° C.
The solution in Reactor 2 was transferred to Reactor 1 over 50 min while controlling the internal temperature of Reactor 1 from 10° C. to 20° C. Once the addition was complete, the jacket temperature was warmed at 20° C. and the resulting mixture was stirred for 16 hr. When UPLC analysis confirmed that the bis-alkylated intermediate was fully formed (target <3% mono-alkylated intermediate), the entire batch was filtered and the filtrate was sent into a clean reactor. The residual TEA-HCl cake was washed with dioxane (300 mL) and the wash was combined with the filtrate. The resulting dioxane solution was then heated to 80° C. and held for 3 hr. After sampling for reaction completion (<1% intermediate remaining), the batch was distilled (pot temp=80° C.) under partial vacuum (400 mbar) to less than half volume. The reaction mixture was diluted with EtOAc (2 L) and washed twice with water (2×2 L). The mixture was then washed with 0.5 N sodium bicarbonate (2 L) and then dried over sodium sulfate (360 g, 2 wt eq) and filtered into a clean dry reactor. The EtOAc solution was concentrated under partial vacuum to about 400 mL total volume resulting in the formation of a thick slurry. The mixture was cooled to 0° C. and stirred for 1 hr and then filtered and washed with cold EtOAc (200 mL) and then dried in a vacuum oven at 40° C. to give 173 g of the title compound. A second crop of product was isolated by concentrating the filtrate and then cooling, granulating and filtering to give an additional 28.4 g of the desired product. In total, the title compound was isolated in 61% yield (201 g, 568 mmol). 1H NMR (400 MHz, DMSO-d6) δ ppm 7.37-7.29 (m, 4 H) 7.29-7.23 (m, 1 H) 5.36 (dd, J=7.3, 3.8 Hz, 1 H) 4.79-4.73 (m, 1 H) 4.48 (d, J=4.8 Hz, 2 H) 3.38-3.31 (m, 2 H), 3.70 (d, J=13.4 Hz, 1 H) 3.62 (d, J=13.4 Hz, 1 H) 3.13-2.99 (m, 2 H) 2.48-2.40 (m, 2 H) 1.46 (s, 9 H). m/z (EI+) for C16H22N2O5S 355.2 (M+H)+.
Preparation Step 4A: Preparation of (3R,4R)-1-benzyl-4-fluoropyrrolidin-3-amine bis-tosylate
A solution of 1M tetrabutylammonium fluoride in THF (1.27 L, 1.27 mol, 2.5 eq) and (3aR,6aS)-5-benzyl-2,2-dioxo-tetrahydro-1-oxa-2λ6-thia-3-5-diaza-pentalene-3-carboxylic acid t-butyl ester (180 g, 0.508 mol, 1.0 eq) were heated at 60° C. (jacket temperature) for 2 hr. Upon reaction completion, the mixture was partially distilled under vacuum to remove the THF. After concentration to a low stir volume, THF was displaced with EtOAc (2×500 mL). After again reducing to a low stir volume, EtOAc (3.6 L) and p-toluenesulfonic acid monohydrate (396 g, 2.10 mol, 4.1 eq) were charged and heated at 80° C. for 2 hr. The mixture was cooled to 10° C. over 1.5 hr and then granulated at 10° C. for 2 hr. The solid product was filtered and washed with EtOAc (2×900 mL) and dried at 50° C. in a vacuum oven for 12 hr. The title compound was isolated as an air stable crystalline solid in 83% yield (231 g, 419 mmol). 1H NMR (400 MHz, D2O) δ ppm 7.69-7.61 (m, 4 H) 7.56-7.42 (m, 5 H) 7.36-7.29 (m, 4 H) 5.65-5.49 (m, 1 H) 4.47 (br. s., 2H) 4.37-4.23 (m, H) 4.15 (ddd, J=12.8, 8.2, 1.4 Hz, 1 H) 3.88 (dd, J=19.1, 1.2 Hz, 1 H), 3.74 (ddd, J=33.2, 14.0, 5.5 Hz, 1 H) 3.44 (dd, J=12.8, 8.2 Hz, 1 H) 2.34 (s, 6 H). m/z (EI+) for C11H15FN2 194.8 (M+H)+.
A suspension of 1,1′-carbonyldiimidazole (73.0 g, 441 mmol, 1.1 eq) in acetonitrile (3.3 L) was stirred at 20° C. until a clear solution was obtained. 3-(methylsulfonyl)propanoic acid (67.0 g, 440 mmol, 1.1 eq) was then added and the mixture was stirred at 25° C. for 3 hr. (3R,4R)-1-benzyl-4-fluoropyrrolidin-3-amine bis-tosylate (220 g, 400 mmol, 1.0 eq) was added and the mixture was stirred at 25° C. for 16 hr resulting in a fine white slurry. The solids were filtered off and the byproduct cake washed with acetonitrile (600 mL). The acetonitrile solution was then concentrated to a low stir volume and then taken up in EtOAc (2.0 L) and washed with 1 N aqueous sodium bicarbonate (1.3 L). The aqueous layer was back extracted with EtOAc (500 mL) and the combined EtOAc layers were washed with water (1.0 L). The resulting EtOAc solution was distilled to remove about 2.0 L of distillate and then displaced with 2-propanol under atmospheric conditions until the internal temperature rose to 78° C. while maintaining a total volume of 2 L. The batch was then cooled to 20° C. and granulated at 20° C. for 12 hr resulting in product crystallization. The desired product was isolated by filtration and the cake washed with 2-propanol (600 mL), then dried in an oven at 40° C. under reduced pressure for 12 hr. The title compound (108 g, 308 mmol) was isolated in 77% yield. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.36 (br. d., J=7.0 Hz, 1 H) 7.37-7.29 (m, 4 H) 7.29-7.23 (m, 1 H) 4.90 (ddt, J=53.4, 5.3, 2×1.7 Hz, 1 H) 4.25 (dddd, J=26.4, 13.9, 7.0, 1.4 Hz, 1 H) 3.61 (d, J=13.2 Hz, 1 H) 3.57 (d, J=13.2 Hz, 1 H) 3.36-3.28 (m, 2 H) 3.03 (dd, J=9.3, 7.5 Hz, 1 H) 2.97 (s, 3 H) 2.80 (dd, J=24.0, 11.6 Hz, 1 H) 2.66 (ddd, J=30.6, 11.6, 5.3 Hz, 1 H) 2.57 (td, 2×7.7, 1.4 Hz, 2 H) 2.18 (dd, J=9.4, 6.7 Hz, 1 H). m/z (EI+) for C15H21FN2O3S 329.7 (M+H)+.
To a Parr reactor was added N-((3R,4R)-1-benzyl-4-fluoropyrrolidin-3-yl)-3-(methylsulfonyl)propanamide (86.5 g, 263 mmol, 1.0 eq), palladium hydroxide (20% on carbon, 2.59 g, 3.69 mmol, 3 wt/wt %) and MeOH (430 mL). The reactor was purged three times with nitrogen (50 psi) and then purged three times with hydrogen (20 psi). The reactor was heated at 50° C. and then pressurized to 50 psi while stirring at 1200 rpm. The material was hydrogenated for 7 hr and then cooled to 20° C. and purged with nitrogen. The mixture was filtered to remove the catalyst and the cake was washed with MeOH (173 mL). The combined filtrate and wash were concentrated to about 200 mL followed by addition of MTBE (200 mL) and then concentrated to a low stir volume. Additional MTBE (200 mL) was added and the resulting slurry granulated at 20° C. for 16 hr. The desired product was isolated by filtration, washed with MTBE (300 mL) and then dried in an oven at 40° C. for 12 hr. The title compound was isolated in 90% yield (53.3 g, 224 mmol) as a white crystalline solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.15 (br. d., J=6.8 Hz, 1 H) 4.96-4.78 (m, 1 H) 4.14-4.01 (m, 1 H) 3.32 (dd, J=8.0, 7.3 Hz, 2 H) 3.13 (dd, J=11.8, 6.8 Hz, 1 H) 3.01-2.93 (m, 1 H) 2.98 (s, 3 H) 2.88 (d, J=3.0 Hz, 1 H) 2.60 (br. s., 1 H) 2.5 7-2.52 (m, 3 H). m/z (EI+) for C8H15FN2O3S 239.1 (M+H)+.
Step 1: Preparation of 2-fluoro-N-(3-methoxy-1-methyl-1H-pyrazol-4-yl)-9H-purin-6-amine
A suspension of 6-chloro-2-fluoro-9H-purine (88% potency, 5.90 kg, 30.20 mol, 1.00 eq), 3-methoxy-1-methyl-1H-pyrazol-4-amine hydrochloride (98% potency, 5.55 kg, 33.22 mol, 1.10 eq), and sodium bicarbonate (10.1 kg, 120.81 mol, 4.00 eq) in EtOAc (106 L) was stirred at 50° C. for 12 hr. The reaction mixture was then cooled to 20° C., granulated for 1 hr, filtered, and the solids were washed with EtOAc (18 L) and dried on the filter. The crude product was charged back into the reactor and suspended in water (106 L) and stirred at 35° C. for 2 hr. The resulting slurry was cooled to 20° C. and the desired product was isolated by filtration and the cake was washed with water (30 L) and then with EtOAc (30 L) and dried for 16 hr at 50° C. to give the title compound (6.26 kg, 23.8 mol, 79% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 13.03 (br. s., 1 H) 9.21 (br. s., 1 H) 8.18 (br. s., 1 H) 7.74 (br. s., 1 H) 3.81 (br. s., 3 H) 3.71 (s, 3 H). m/z (APCI+) for C10H11FN7O 264.2 (M+H)+.
Step 2: Preparation of 2-fluoro-N-(3-methoxy-1-methyl-1H-pyrazol-4-yl)-9-methyl-9H-purin-6-amine
To a 100 L reactor fitted with a caustic scrubber was added 2-methyltetrahydrofuran (44.0 L), 2-fluoro-N-(3-methoxy-1-methyl-1H-pyrazol-4-yl)-9H-purin-6-amine (2.20 kg, 8.36 mol, 1.00 eq) and potassium phosphate tribasic (7.10 kg, 33.43 mol mmol, 4.00 eq). The resulting mixture was stirred at 5° C. and dimethyl sulfate (1.42 kg, 11.28 mol, 1.35 eq) was added and the resulting mixture was stirred at 5° C. for 1 hr. The reaction was warmed from 5° C. to 15° C. over 2 hr and then held at 15° C. for 20 hr. The reaction mixture was cooled to 5° C. and quenched with water (44.0 L) while maintaining the internal temperature below 10° C. The mixture was then heated at 50° C. for 2 hr and then cooled to 10° C. and granulated for 2 hr. The product was isolated by filtration and washed with water (11.0 L) and then with 2-methyltetrahydrofuran (11.0 L). The cake was dried under vacuum at 40° C. for 8 hr to give the title compound (1.99 kg, 7.18 mol, 86% yield) as an off white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.23 (br. s., 1 H) 8.13 (br. s., 1 H) 7.67 (s, 1 H) 3.78 (s, 3 H)3.70 (s, 3 H) 3.69 (br. s., 3 H). m/z (APCI+) for C11H13FN7O 278.2 (M+H)+.
Step 3: Preparation of N-((3R,4R)-4-fluoro-1-(6-((3-methoxy-1-methyl-1H-pyrazol-4-yl)amino)-9-methyl-9H-purin-2-yl)pyrrolidin-3-yl)acrylamide
To a 200 L Hastelloy reactor heated to 40° C. was added sulfolane (22.4 L) and N-((3R,4R)-4-fluoropyrrolidin-3-yl)-3-(methylsulfonyl)propanamide (4.03 kg, 16.9 mol, 1.05 eq) and stirred the resulting mixture until all solids were dissolved. To this solution was added 2-fluoro-N-(3-methoxy-1-methyl-1H-pyrazol-4-yl)-9-methyl-9H-purin-6-amine (4.47 kg, 16.1 mol, 1.00 eq) and N,N-diisopropylethylamine (8.50 L, 48.7 mol, 3.0 eq) and the mixture heated at 115° C. for 16 hr. The reaction mixture was cooled to 30° C., and a solution of potassium hydroxide (2.26 kg, 40.3 mol, 2.5 eq) in water (44.7 L) was added. After stirring for 4 hr, the reaction mixture was cooled to 20° C., water (44.7 L) was added and the resulting mixture granulated for 12 hr. The crude product was isolated on a Nutsche filter and washed with water (27 L) and then dried under nitrogen on the filter. The reactor was cleaned and then charged with water (35.8 L) and acetone (53.6 L). The crude product cake was charged back into the reactor and heated to 60° C. until all of the solids had dissolved. The batch was then cooled to 40° C. and then transferred into a speck free 100 L reactor via an in-line 10 μm filter. The 200 L reactor, line and filter were rinsed with acetone (5 L) and sent into the 100 L reactor. The batch was concentrated with the jacket temperature set at 70° C. under partial vacuum until the acetone content reduced to 5 wt %, as determined by gas chromatography head space. The batch was then cooled to 20° C. and granulated for 4 hr. The product was filtered, washed with water (18 L) and dried in a vacuum oven at 55° C. for 8 hr. The title compound (3.942 kg, 9.49 mol, 59%) was isolated as a white crystalline solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.44 (d, J=6.5 Hz, 1 H) 7.97 (s, 1 H) 7.82 (s, 1 H) 7.78 (s, 1 H) 6.23 (dd, J=10.0, 17.0 Hz, 1 H) 6.14 (dd, J=2.8, 17.0 Hz, 1 H) 5.62 (dd, J=2.8, 10.0 Hz, 1 H) 5.12 (d, J=51.0 Hz, 1 H) 4.46 (td, J=6.0, 11.9 Hz, 1 H) 3.88-3.6 (m, 4 H) 3.82 (s, 3 H) 3.71 (s, 3 H) 3.62 (s, 3 H). m/z (APCI+) for C18H23FN9O2 416.3 (M+H)+.
Summary of 1st generation and 2nd generation EGFR inhibitors
REFERENCES
Planken, S.; Murray, B. W.; Lafontaine, J.; Weinrich, S.; Hemkens, M.; Kath, J. C.; Nair, S. K.; Johnson, T. O.; Cheng, H.; Sutton, S. C.; Zientek, M.; Yin, M. -J.; Solowiej, J.; Nagata, A.; Gajiwala, K. Abstracts of Papers, 249th ACS National Meeting & Exposition, Denver, CO, United States, March 22–26, 2015; MEDI-248
Programmed Cell Death 1 or PD-1 (also referred to as PDCD1) is a 50 to 55 kD type I membrane glycoprotein (Shinohara T et al, Genomics, 1994, Vol. 23, No. 3, pp. 704-706). PD-1 is a receptor of the CD28 superfamily that negatively regulates T cell antigen receptor signalling by interacting with the specific ligands and is suggested to play a role in the maintenance of self tolerance.
PD-1 peptide relates to almost every aspect of immune responses including autoimmunity, tumour immunity, infectious immunity, transplantation immunity, allergy and immunological privilege.
The PD-1 protein’s structure comprise of—
an extracellular IgV domain followed by
a transmembrane region and
an intracellular tail
The intracellular tail contains two phosphorylation sites located in an immunoreceptor tyrosine-based inhibitory motif and an immunoreceptor tyrosine-based switch motif, which suggests that PD-1 negatively regulates TCR signals. Also, PD-1 is expressed on the surface of activated T cells, B cells, and macrophages, (Y. Agata et al., Int Immunol 8, 765, May 1996) suggesting that compared to CTLA-4 ((Cytotoxic T-Lymphocyte Antigen 4, also known as CD152 (Cluster of differentiation 152) is a protein that also plays an important regulatory role in the immune system), PD-1 more broadly negatively regulates immune responses.
PD-1 has two ligands, PD-L1 (Programmed Death Ligand for PDCD1L1 or B7-H1) (Freeman G J et al, Journal of Experimental Medicine, 2000, Vol. 19, No. 7, pp. 1027-1034) and PD-L2 (Programmed Death Ligand 2 or PDCD1L2 or B7-DC) (Latchman Y et al, Nature Immunology, 2001, Vol. 2, No. 3, pp. 261-267), which are members of the B7 family. PD-L1 is known to be expressed not only in immune cells, but also in certain kinds of tumour cell lines (such as monocytic leukaemia-derived cell lines, mast cell tumour-derived cell lines, hematoma-derived cell lines, neuroblastoma-derived cell lines, and various mammary tumour-derived cell lines) and in cancer cells derived from diverse human cancer tissues (Latchman Y et al, Nature Immunology, 2001, Vol. 2, No. 3, pp. 261-267) and on almost all murine tumour cell lines, including PA1 myeloma, P815 mastocytoma, and B16 melanoma upon treatment with IFN-γ (Y. Iwai et al., Proc Natl Acad Sci USA 99, 12293, Sep. 17, 2002 and C. Blank et al., Cancer Res 64, 1140, February, 2004). Similarly PD-L2 expression is more restricted and is expressed mainly by dendritic cells and a few tumour cell lines. PD-L2 expression has been verified in Hodgkin’s lymphoma cell lines and others. There is a hypothesis that some of the cancer or tumour cells take advantage from interaction between PD-1 and PD-L1 or PD-L2, for suppressing or intercepting T-cell immune responses to their own (Iwai Y et al, Proceedings of the National Academy of Science of the United States of America, 2002, Vol. 99, No. 19, pp. 12293-12297).
Tumour cells and virus (including HCV and HIV) infected cells are known to express the ligand for PD-1 (to create Immunosuppression) in order to escape immune surveillance by host T cells. It has been reported that the PD-1 gene is one of genes responsible for autoimmune diseases like systemic lupus erythematosis (Prokunina et al, Nature Genetics, 2002, Vol. 32, No. 4, 666-669). It has also been indicated that PD-1 serves as a regulatory factor for the onset of autoimmune diseases, particularly for peripheral self-tolerance, on the ground that PD-1-deficient mice develop lupus autoimmune diseases, such as glomerulonephritis and arthritis (Nishimura H et al, International Immunology, 1998, Vol. 10, No. 10, pp. 1563-1572; Nishimura H et al, Immunity, 1999, Vol. 11, No. 2, pp. 141-151), and dilated cardiomyopathy-like disease (Nishimura H et al, Science, 2001, Vol. 291, No. 5502, pp. 319-332).
Hence, in one approach, blocking the interaction of PD-1 with its ligand (PD-L1, PD-L2 or both) may provide an effective way for specific tumour and viral immunotherapy.
Wood et al in U.S. Pat. No. 6,808,710 discloses method for down modulating an immune response comprising contacting an immune cell expressing PD-1 with an antibody that binds to PD-1, in multivalent form, such that a negative signal is transduced via PD-1 to thereby down modulate the immune response. Such an antibody may be a cross-linked antibody to PD-1 or an immobilized antibody to PD-1.
Freeman et al in U.S. Pat. No. 6,936,704 and its divisional patent U.S. Pat. No. 7,038,013 discloses isolated nucleic acids molecules, designated B7-4 nucleic acid molecules, which encode novel B7-4 polypeptides, isolated B7-4 proteins, fusion proteins, antigenic peptides and anti-B7-4 antibodies, which co-stimulates T cell proliferation in vitro when the polypeptide is present on a first surface and an antigen or a polyclonal activator that transmits an activating signal via the T-cell receptor is present on a second, different surface.
There are some reports regarding substances inhibiting immunosuppressive activity of PD-1, or interaction between PD-1 and PD-L1 or PD-L2, as well as the uses thereof. A PD-1 inhibitory antibody or the concept of a PD-1 inhibitory peptide is reported in WO 01/14557, WO 2004/004771, and WO 2004/056875. On the other hand, a PD-L1 inhibitory antibody or a PD-L1 inhibitory peptide is reported in WO 02/079499, WO 03/042402, WO 2002/086083, and WO 2001/039722. A PD-L2 inhibitory antibody or a PD-L2 inhibitory peptide is reported in WO 03/042402 and WO 02/00730.
WO2007005874 describes isolated human monoclonal antibodies that specifically bind to PD-L1 with high affinity. The disclosure provides methods for treating various diseases including cancer using anti-PD-L1 antibodies.
US2009/0305950 describes multimers, particularly tetramers of an extracellular domain of PD-1 or PD-L1. The application describes therapeutic peptides.
Further, the specification mentions that peptides can be used therapeutically to treat disease, e.g., by altering co-stimulation in a patient. An isolated B7-4 or PD-1 protein, or a portion or fragment thereof (or a nucleic acid molecule encoding such a polypeptide), can be used as an immunogen to generate antibodies that bind B7-4 or PD-1 using standard techniques for polyclonal and monoclonal antibody preparation. A full-length B7-4 or PD-1 protein can be used, or alternatively, the invention provides antigenic peptide fragments of B7-4 or PD-1 for use as immunogens. The antigenic peptide of B7-4 or PD-1 comprises at least 8 amino acid residues and encompasses an epitope of B7-4 or PD-1 such that an antibody raised against the peptide forms a specific immune complex with B7-4 or PD-1. Preferably, the antigenic peptide comprises at least 10 amino acid residues, more preferably at least 15 amino acid residues, even more preferably at least amino acid residues, and most preferably at least 30 amino acid residues.
Freeman et al in U.S. Pat. No. 7,432,059 appears to disclose and claim methods of identifying compounds that up modulate T cell activation in the presence of a PD-1-mediated signal. Diagnostic and treatment methods utilizing compositions of the invention are also provided in the patent.
Further, Freeman et al in U.S. Pat. No. 7,709,214 appears to cover methods for up regulating an immune response with agents that inhibit the interactions between PD-L2 and PD-1.
Despite existence of many disclosures as discussed above, however, a significant unmet medical need still exists due to the lack of effective peptides or modified peptides as therapeutic agents as alternatives in the therapeutic area. It is known that synthetic peptides offer certain advantages over antibodies such as ease of production with newer technologies, better purity and lack of contamination by cellular materials, low immunogenicity, improved potency and specificity. Peptides may be more stable and offer better storage properties than antibodies. Moreover, often peptides possess better tissue penetration in comparison with antibodies, which could result in better efficacy. Peptides can also offer definite advantages over small molecule therapeutics counterparts such as lesser degree of toxicity and lower probability of drug-drug interaction.
The present invention therefore may provide the solution for this unmet medical need by offering novel synthetic peptide and its derivatives which are based on the PD1 ectodomain.
Aurigene team: (from left) Brahma Reddy V, Thomas Antony, Murali Ramachandra, Venkateshwar Rao G, Wesley Roy Balasubramanian, Kishore Narayanan, Samiulla DS, Aravind AB, and Shekar Chelur
SNTSESFK(SNTSESF)FRVTQLAPKAQIKE-NH2 (SEQ ID NO: 49)
Example 2 Synthesis of
Synthesis of Linear Fragment—Fmoc-FRVTQLAPKAQIKE
Desiccated CLEAR-Amide resin ((100-200 mesh) 0.4 mmol/g, 0.5 g) was distributed in 2 polyethylene vessels equipped with a polypropylene filter. The linear peptide synthesis on solid phase were carried out automatically, using Symphony parallel synthesizer (PTI) using the synthesis programs mentioned in the table below. Swelling, C-terminal amino acid [Fmoc-Glu(OtBu)-OH] attachment and capping of the peptidyl resin was carried out as per the protocol in Table I. Subsequent amino acid coupling was carried out as mentioned in Table II. The amino acids used in the synthesis were Fmoc Phe-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Thr(OtBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Leu-OH, Fmoc-Ala-OH, Fmoc-Pro-OH, Fmoc-Ile-OH. After the completion of Fmoc-Phe-OH coupling the resin was taken out form peptide synthesiser and manual coupling was carried out as follows
Fmoc-Phe-OH peptidyl resin from automated synthesiser was pooled in to a glass vessel with frit. The Fmoc group of the peptidyl resin was deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (10 m L). The resin was washed with DMF (6×15 m L), DCM (6×15 m L) and DMF (6×15 m L). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive. Fmoc-Lys (Fmoc)-OH (0.48 g; 4 equiv. 0.8 m mol) in dry DMF was added to the deprotected resin and coupling was initiated with DIC (0.15 m L; 5 equiv, 1 m mol) and HOBT (0.08 g; 5 equiv, 0.6 m mol) in DMF. The concentration of each reactant in the reaction mixture was approximately 0.4 M. The mixture was rotated on a rotor at room temperature for 3 h. Resin was filtered and washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of coupling was negative. The Fmoc group on the peptidyl resin is deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (15 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive. After the deprotection of Fmoc group on Fmoc-Lys(Fmoc)-attached peptidyl resin the peptide chain growth was carried out from both the free amino terminus suing 8 equivalent excess of amino acid (1.6 m mol, 8 equivalent excess of HOBt (0.22 g, 1.6 m mol) and 10 equivalent excess of DIC (0.32 m L, 2 m mol) relative to resin loading. The coupling was carried out at room temperature for 3 h. The amino acids coupled to the peptidyl resin were; Fmoc-Phe-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Glu (OtBu)-OH (0.68 g; 8 equiv, 1.6 m mol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 m mol), Fmoc-Thr (OtBu)-OH (0.64 g; 8 equiv, 1.6 m mol), Fmoc-Asn (Trt)-OH (0.95 g; 8 equiv, 1.6 m mol) and N-terminus amino acids as Boc-Ser (OtBu)-OH (0.41 g; 8 equiv, 1.6 m mol) The peptidyl resin was cleaved as mentioned in procedure for cleavage using cleavage cocktail A to yield (565 mg), 70% yield. The crude material was purified by preparative HPLC on Zorbax Eclipse XDB-C18 column (9.4 mm×250 mm, 5 μm) with buffer A: 0.1% TFA/Water, buffer B: Acetonitrile. The peptide was eluted by gradient elution 0-5 min=5-10% buffer B, 10-20 min=29% buffer B with a flow rate of 7 mL/min. HPLC: (method 1): RT-12 min (96%); LCMS Calculated Mass: 3261.62, Observed Mass: 1631.6 [M/2+H]+; 1088 [M/3+H]+); 816.2[M/4+H]+;
Compound 8 (SEQ ID NO: 49) SNTSESFK(SNTSESF)FRVTQLAPKAQIKE-NH2
Example 2Synthesis of Sequence Shown in SEQ ID NO: 49
Synthesis of Linear Fragment—Fmoc-FRVTQLAPKAQIKE
Desiccated CLEAR-Amide resin ((100-200 mesh) 0.4 mmol/g, 0.5 g) was distributed in 2 polyethylene vessels equipped with a polypropylene filter. The linear peptide synthesis on solid phase were carried out automatically, using Symphony parallel synthesizer (PTI) using the synthesis programs mentioned in the table below. Swelling, C-terminal amino acid [Fmoc-Glu(OtBu)-OH] attachment and capping of the peptidyl resin was carried out as per the protocol in Table I. Subsequent amino acid coupling was carried out as mentioned in Table II. The amino acids used in the synthesis were Fmoc Phe-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Val-OH, Fmoc-Thr(OtBu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Leu-OH, Fmoc-Ala-OH, Fmoc-Pro-OH, Fmoc-Ile-OH. After the completion of Fmoc-Phe-OH coupling the resin was taken out form peptide synthesiser and manual coupling was carried out as follows.
Fmoc-Phe-OH peptidyl resin from automated synthesiser was pooled in to a glass vessel with frit. The Fmoc group of the peptidyl resin was deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (10 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive.
Fmoc-Lys (Fmoc)-OH (0.48 g; 4 equiv. 0.8 mmol) in dry DMF was added to the deprotected resin and coupling was initiated with DIC (0.15 mL; 5 equiv, 1 mmol) and HOBT (0.08 g; 5 equiv, 0.6 mmol) in DMF. The concentration of each reactant in the reaction mixture was approximately 0.4 M. The mixture was rotated on a rotor at room temperature for 3 h. Resin was filtered and washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of coupling was negative. The Fmoc group on the peptidyl resin is deprotected by treating it twice with 20% (v/v) piperidine/DMF solution for 5 and 15 min (15 mL). The resin was washed with DMF (6×15 mL), DCM (6×15 mL) and DMF (6×15 mL). Kaiser test on peptide resin aliquot upon completion of Fmoc-deprotection was positive.
After the deprotection of Fmoc group on Fmoc-Lys(Fmoc)-attached peptidyl resin the peptide chain growth was carried out from both the free amino terminus suing 8 equivalent excess of amino acid (1.6 mmol, 8 equivalent excess of HOBt (0.22 g, 1.6 mmol) and 10 equivalent excess of DIC (0.32 mL, 2 mmol) relative to resin loading. The coupling was carried out at room temperature for 3 h. The amino acids coupled to the peptidyl resin were; Fmoc-Phe-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Glu (OtBu)-OH (0.68 g; 8 equiv, 1.6 mmol), Fmoc-Ser (OtBu)-OH (0.62 g; 8 equiv, 1.6 mmol), Fmoc-Thr (OtBu)-OH (0.64 g; 8 equiv, 1.6 mmol), Fmoc-Asn (Trt)-OH (0.95 g; 8 equiv, 1.6 m mol) and N-terminus amino acids as Boc-Ser (OtBu)-OH (0.41 g; 8 equiv, 1.6 mmol) The peptidyl resin was cleaved as mentioned in procedure for cleavage using cleavage cocktail A to yield (565 mg), 70% yield. The crude material was purified by preparative HPLC on Zorbax Eclipse XDB-C18 column (9.4 mm×250 mm, 5 μm) with buffer A: 0.1% TFA/Water, buffer B:Acetonitrile. The peptide was eluted by gradient elution 0-5 min=5-10% buffer B, 10-20 min=29% buffer B with a flow rate of 7 mL/min. HPLC: (method 1): RT—12 min (96%); LCMS Calculated Mass: 3261.62, Observed Mass: 1631.6 [M/2+H]+; 1088 [M/3+H]+😉; 816.2[M/4+H]+.
Aurigene and Pierre Fabre Pharmaceuticals Announce a Licensing Agreement for a New Cancer Therapeutic in Immuno-oncology: AUNP12, an Immune Checkpoint Modulator Targeting the PD-1 Pathway
Pierre Fabre are thus reinforcing their oncology portfolio which already enjoys a combination of chemotherapies, monoclonal antibodies and immuno-conjugates assets at various development phases
CASTRES, France and BANGALORE, India, February 13, 2014 /PRNewswire/ —
Pierre Fabre, the third largest French pharmaceutical company, and Aurigene, a leading biotech company based in India, today announced that the two companies have entered into a collaborative license, development and commercialization agreement granting Pierre Fabre global Worldwide rights (excluding India) to a new immune checkpoint modulator, AUNP-12.
AUNP-12 offers a breakthrough mechanism of action in the PD-1 pathway compared to other molecules currently in development in the highly promising immune therapy cancer space. AUNP-12 is the only peptide therapeutic in this pathway and could offer more effective and safer combination opportunities with emerging and established treatment regimens. AUNP-12 will be in development for numerous cancer indications.
Under the terms of this agreement, Aurigene will receive an upfront payment from Pierre Fabre. Aurigene will also receive additional milestone payments based upon the continued development, regulatory progresses and commercialization of AUNP-12.
“We are pleased that Pierre Fabre see the PD-1 program as a strategic asset in their portfolio. Overall, the deal structure, in line with the financial terms that have been seen in this space, demonstrate the importance that Pierre Fabre attach to the program,” said CSN Murthy, CEO, Aurigene.
“The plans that Pierre Fabre have detailed for the development of this differentiated asset highlight the long-term opportunities for this novel cancer therapeutic,” added Murali Ramachandra, Sr VP, Research, Aurigene.
“This agreement, in the field of oncology, is fully consistent with our vision to build Pierre Fabre’s future in prescription drugs, from a combination of cutting-edge internal R&D capabilities and license partnerships with innovative biotech companies like Aurigene,” stated Bertrand Parmentier, CEO, Pierre Fabre.
“With this deal, Pierre-Fabre Pharmaceuticals are reinforcing their portfolio of oncology assets and capitalizing on their proven capabilities in developing biological compounds such as monoclonal antibodies and immuno-conjugates. We have been impressed by the science at Aurigene and encouraged by the differentiated profile reported for AUNP-12,” added Frédéric Duchesne, President, Pierre Fabre Pharmaceuticals.
About immuno-oncology
Immuno-oncology is an emerging field in cancer therapy, where the body’s own immune system is harnessed to fight against cancer. This approach of targeting cancer through immune response has had a breakthrough when robust and sustained responses were obtained only upon blocking the immune checkpoint targets (such as PD-1 and CTLA4). Recent successes in clinical trials performed with such therapies suggest that immunotherapy should be considered alongside surgery, chemotherapy, radiotherapyand targeted therapyas the fifth cornerstone of cancer treatment.
PD-1 (Programmed cell Death 1) is a receptor that negatively regulates T-cell activation by interacting with specific ligands PD-L1 and PD-L2. Tumor cells express these ligands and thereby escape from the action of T-cells.
About AUNP-12
AUNP-12is a branched 29-amino acid peptide sequence engineered from the PD-L1/ L2 binding domain of PD-1It blocks the PD-1/PD-L1, PD-1/PD-L2 and PD-L1/CD80 pathways.AUNP-12 is highly effective in antagonizing PD-1 signaling, with desirable in vivo exposure upon subcutaneous dosing. It inhibits tumor growth and metastasis in preclinical models of cancer and is well tolerated with no overt toxicity at any of the tested doses.
About Aurigene
Aurigene is a biotech focused on development of innovative small molecule and peptide therapeutics for Oncology and Inflammation; key focus areas for Aurigene are Immuno-oncology, Epigenetics and the Th17 pathway.Aurigene’s PD-1 program is the first of several peptide-based immune checkpoint programs that are at different stages of Discovery.
Aurigene has partnered with several big pharma and mid-pharma companies in the US and Europe, and has delivered multiple clinical compounds through these partnerships. With over 500 scientists, Aurigene has collaborated with 6 of the top 10 pharma companies.
Aurigene’s pre-clinical pipeline includes (1) Selective and pan-BET Bromodomain inhibitors (2) RoR gamma reverse agonists (3) EZH2 inhibitors (4) NAMPT inhibitors and (5) Several immune check point peptide inhibitor programs.
Pierre Fabre is a privately-owned health care company created in 1961 by Mr Pierre Fabre. It is the second largest French independent pharmaceutical group with 2013 sales amounting to about €2 billion (yet to be audited) across 140 countries. The company is structured around two divisions: Pharmaceuticals (Prescription drugs, OTC, Oral care) and Dermo-cosmetics. Prescription drugs are organized around four main franchises: oncology, dermatology, women’s health and neuropsychiatry. Pierre Fabre employs some 10000 people worldwide, including1 300 inR&D. The company allocates about 20% of its pharmaceuticals sales to R&D and relies on more than 25 years of experience in the discovery, development and global commercialization of innovative drugs in oncology. Pierre Fabre has a long commitment to oncology and immunology with major R&D centers in France: the Pierre Fabre immunology Centre (CIPF) in Saint Julien en Genevois and the Pierre Fabre Research Institute (IRPF) located on the Toulouse-Oncopole campus which has been officially recognized as a National Center of Excellence for cancer research since 2012.
P. Sasikumar, R. Shrimali, S. Adurthi, R. Ramachandra, L. Satyam, A. Dhudashiya, D. Samiulla, K. B. Sunilkumar and M. Ramachandra, “A novel peptide therapeutic targeting PD1 immune checkpoint with equipotent antagonism of both ligands and a potential for better management of immune-related adverse events,” Journal for ImmunoTherapy of Cancer, vol. 1, no. Suppl 1, O24, 2013.
P. G. N. Sasikumar, M. Ramachandra, S. K. Vadlamani, K. R. Vemula, L. K. Satyam, K. Subbarao, K. R. Shrimali and S. Kandepudu (Aurigene Discovery Technologies Ltd, Bangalore, India), “Immunosuppression modulating compounds”, US Patent application US 2011/0318373, 29 Dec 2011.
P. G. Sasikumar, L. K. Satyam, R. K. Shrimali, K. Subbarao, R. Ramachandra, S. Vadlamani, A. Reddy, A. Kumar, A. Srinivas, S. Reddy, S. Gopinath, D. S. Samiulla and M. Ramachandra, “Demonstration of anti-tumor efficacy in multiple preclinical cancer models using a novel peptide inhibitor (Aurigene-012) of the PD1 signaling pathway,” Cancer Research, vol. 72, no. 8 Suppl. 1, Abstract 2850, 2012.
P. G. N. Sasikumar, M. Ramachandra, S. K. Vadlamani, K. R. Shrimali and K. Subbarao, “Therapeutic compounds for immunomodulation” (Aurigene Discovery Technologies Ltd, Bangalore, India), PCT Patent Application WO 2012/168944, 13 Dec 2012.
P. G. N. Sasikumar and M. Ramachandra, “Immunomodulating cyclic compounds from the BC loop of human PD1” (Aurigene Discovery Technologies Ltd, Bangalore, India), PCT Patent Application WO/2013/144704, 3 Oct 2013.
P. G. N. Sasikumar, M. Ramachandra and S. S. S. Naremaddepalli, “Peptidomimetic compounds as immunomodulators” (Aurigene Discovery Technologies Ltd, Bangalore, India), US Patent Application US 2013/0237580, 12 Sep 2013.
A. H. Sharpe, M. J. Butte and S. Oyama (Harvard College), “Modulators of immunoinhibitory receptor PD-1, and methods of use thereof”, PCT Patent Application WO/2011/082400, 7 Jul 2011.
Mr. CSN Murthy began his career with ICICI Ventures, India’s first Venture Capital fund. He was subsequently a management consultant to the Pharma and Chemical sectors. Later, he worked in the Business Development and General Management functions in Pharmaceutical companies, including as the Chief Operating Officer of Gland Pharma Ltd. CSN holds a Bachelors degree in Chemical Engineering from the Indian Institute of Technology (IIT), Madras and an MBA from the Indian Institute of Management (IIM), Bangalore.
Dr.Thomas Antony
Associate Research Director, Aurigene Discovery Technologies Ltd.
Dr.Thomas Antony did his Ph.D in Biophysical Chemistry from University of Delhi and had his postdoctoral training at Jawaharlal Nehru University- Delhi, The University of Medicine and Dentistry of New Jersey- USA, and Max Planck Institute for Biophysical Chemistry- Germany. He is the recipient of many research fellowships, including Max Planck Fellowship and Humboldt Research Fellowship. He has more than 20 years of research experience. Dr.Thomas has published 24 research papers and he is the co-author of three international patents. His core area of expertise is in assay development and screening. At Aurigene, Dr.Thomas leads the Biochemistry and Structural Biology Divisions. He was the coordinator of Aurigene-University of Malaya collaboration programs.
Dr. Kavitha Nellore
Associate Research Director, Aurigene Discovery Technologies Ltd.
Dr. Kavitha Nellore obtained her PhD in Bioengineering from Pennsylvania State University, USA. During this time, she was a fellow of the Huck’s Institute of Life Sciences specializing in Biomolecular Transport Dynamics. She has been at Aurigene for more than a decade, and is currently leading a group of cell biologists at both Bangalore and Kuala Lumpur. At Aurigene, she leads multiple drug discovery programs in the therapeutic areas of inflammation, oncology and immuno-oncology. She plays a key role in target selection as well as validation efforts to add to Aurigene’s pipeline. Kavitha also played a key role in coordinating the Aurigene-University of Malaya collaboration.
To print: Click hereor Select File and then Print from your browser's menu
“Strategic partnerships will boost drug discovery activities in India”
Wednesday, May 16, 2007 08:00 IST
The Bangalore-based Aurigene Discovery Technologies Limited, an independent subsidiary of Dr Reddy’s, is now competing in a mature contract research organization space in the country. Established in 2003, Aurigene Discovery Technologies is a partnership focused collaborative discovery organisation. CSN Murthy is chief executive officer of the company. In an interaction with Nandita Vijay, Murthy cut a clear picture of the contract research scene. Excerpts:
What are the factors led to the scores of partnerships in research and drug discovery space?
India is a cost-effective destination but there is lack of expertise, as it has so far not witnessed a complete cycle of discovery, development and marketing of a compound. Therefore, the possible solution is to enter into strategic business partnerships to combine low costs and excellent global skills. Such alliances make a lot of business sense, because no Indian company can afford to invest in resources to research on clinical development of compounds or identification of new drug targets, which is a highly risky area. The Indian CROs have so far focused largely on chemistry-based projects. But of late, there has been some attempt by CROs to offer biology services. The chemistry services have taken off well and there could be at least 2,000 chemists working in Indian CROs. But the biology service is still in the incipient stage.
Biology demands expertise in areas such as DMPK, cell and assay biology and vivo expertise, which is not as common as chemistry expertise in India. There is also a paradigm shift in assignments for CROs, as they are gearing up to gain higher value from existing projects, including Intellectual Property (IP) generation. Probably, this is where Aurigene has a head start over other CROs. Although we are in research services, our success is in IP generated work. We work with world-class companies like Novo Nordisk, Rheoscience, Debiopharm, Forest Labs and Merck Serono on integrated discovery projects.
What do you think is the uniqueness of Aurigene that sets it apart from other companies?
We are the oldest and know the ins and outs of the Indian pharmaceutical market. Our strengths include modern infrastructure and resourceful, talented scientific pool. Right now, we are working with Forrest Labs to meet the first milestone. We are adding more customers and there is value-addition to projects from existing customers. We are also able to add more customers in the area of drug discovery. This has led to expansion in infrastructure and manpower.
It is understood that a major chunk of the business for CROs is from global customers. What is the reason for this?
That is true. Even Aurigene is not favoring Indian customers. The situation is similar to the information technology sector in the country where local giants like Wipro and Infosys augmented businesses through global partnerships. However, after two decades, they are now serving Indian customers. The same thing will happen in India in the CROs space. Right now, I doubt, if any Indian pharma or biotech company will look for collaboration or business development with Indian counterparts. It makes more business sense to work for global customers.
The whole concept of drug discovery outsourcing business has caught on in the last 18-month. Could you give us an overview of the market?
The reason for the sudden interest in drug discovery is the cost arbitrage. There is downward pressure on prices and upward pressure on costs. Globally, the market size for drug discovery is around $8 billon, excluding the licensing costs. I feel the overall market expansion is not going to happen in terms of increased research spends. The opportunities will come from shift in spend to India from Europe and US. The amount of outsourced value will actually increase substantially in the next few years. In the western countries, pharma giants are busy partnering with biotechnology companies to license early/late-stage compounds. In other words, the biotech companies offer novel technology platforms. In a situation such as this, while the former gets the license rights, the latter earn milestone payments and royalties.
In India, CROs operate differently. The innovation driven companies are taking on ‘collaborative drug discovery’ projects, leveraging the mutual strengths each has to offer the other (cost and expertise respectively). Therefore, the complexion of business is different from that of the Western countries. We would see large outsourcing companies positioning themselves as offshore partners for pharma-biotech majors. They could also go in for a BOT (Build Operate Transfer) model. Under this model, companies will set-up the facility and manage it to make it a ‘centre of excellence’, focusing on certain disease segments. Another option is that some companies can take up their own programmes to develop compounds and enter into licensing partnerships. This is similar to the US and Europe biotech style of working. Both of these practices are likely to happen in India.
How much do you acknowledge science and strategy in Aurigene’s growth?
Science will translate into good value if there is an appropriate business direction to it. The same is true of Aurigene. We are adding value by generating IPs, because it does not need massive infusion of manpower. It definitely costs less to do a discovery in Bangalore than in Boston. Although time taken to generate an IP is higher, it can create potential revenues.
What according to you is the future of the drug discovery sector?
It is at an interesting stage. There is lot of scope for large and medium cap companies to enter India. Pfizer has a large presence in Mumbai for clinical and analytical work. Novartis is setting up a new facility in Hyderabad to focus more on clinical development and analytics. Even Wyeth is reportedly moving towards an expanded relationship with its Indian partners. Bristol Myers Squib has already teamed up with Biocon’s subsidiary Syngene. Companies like Aurigene are gaining visibility. There has never been such a huge interest towards drug discovery partnerships. New players like Advinus and Jubilant have set-up large facilities in Bangalore and Pune, respectively.
/////////AUNP-12, Aurigene, Pierre Fabre Pharmaceuticals, Licensing Agreement, New Cancer Therapeutic, Immuno-oncology, AUNP 12, Immune Checkpoint Modulator Targeting the PD-1 Pathway, PEPTIDES
MS m/z 508.4 [M + H]+. Anal. (C30H33N7O·1.0H2O) C, H, N.
GSK1070916 is a reversible and ATP-competitive inhibitor of Aurora B/C with IC50 of 3.5 nM/6.5 nM; displays >100-fold selectivity against the closely related Aurora A-TPX2 complex(IC50=490 nM).
NMI-900, an Aurora B/C kinase inhibitor, is under development at Cancer Research Technology in phase I/II clinical studies for the treatment of advanced and/or metastatic solid tumors. Other phase I clinical trials for the treatment of solid tumors had been previously completed, in a collaboration between GlaxoSmithKline and Cancer Research Technology, under the Cancer Research UK’s Clinical Development Partnerships (CDP) program.
The drug was originated by GlaxoSmithKline. The rights of the product were acquired by Cancer Research Technology from GlaxoSmithKline after the company elected not to take the program forward. In December 2015, the product was licensed by Cancer Research Technology to Nemucore Medical Innovations for the exclusive worldwide development and commercialization for the treatment of difficult-to-treat cancers.
GlaxoSmithKline, 1250 South Collegeville Road, Collegeville, Pennsylvania 19426
§ Tsukuba Research Laboratories, Japan
J. Med. Chem., 2010, 53 (10), pp 3973–4001
DOI: 10.1021/jm901870q
The Aurora kinases play critical roles in the regulation of mitosis and are frequently overexpressed or amplified in human tumors. Selective inhibitors may provide a new therapy for the treatment of tumors with Aurora kinase amplification. Herein we describe our lead optimization efforts within a 7-azaindole-based series culminating in the identification of GSK1070916 (17k). Key to the advancement of the series was the introduction of a 2-aryl group containing a basic amine onto the azaindole leading to significantly improved cellular activity. Compound 17k is a potent and selective ATP-competitive inhibitor of Aurora B and C with Ki* values of 0.38 ± 0.29 and 1.5 ± 0.4 nM, respectively, and is >250-fold selective over Aurora A. Biochemical characterization revealed that compound 17k has an extremely slow dissociation half-life from Aurora B (>480 min), distinguishing it from clinical compounds 1 and 2. In vitro treatment of A549 human lung cancer cells with compound 17k results in a potent antiproliferative effect (EC50 = 7 nM). Intraperitoneal administration of 17k in mice bearing human tumor xenografts leads to inhibition of histone H3 phosphorylation at serine 10 in human colon cancer (Colo205) and tumor regression in human leukemia (HL-60). Compound 17k is being progressed to human clinical trials.
GSK1070916 is a reversible and ATP-competitive inhibitor of Aurora B/C with IC50 of 3.5 nM/6.5 nM; displays >100-fold selectivity against the closely related Aurora A-TPX2 complex(IC50=490 nM). IC50 Value: 3.5 nM(Aurora B); 6.5 nM(Aurora C) Target: Aurora B/C in vitro: GSK1070916 selectively inhibits Aurora B and Aurora C with Ki of 0.38 nM and 1.5 nM over Aurora A with Ki of 490 nM. Inhibition of Aurora B and Aurora C is time-dependent, with an enzyme-inhibitor dissociation half-life of >480 min and 270 min respectively. In addition, GSK1070916 is also a competitive inhibitor with respect to ATP. Human tumor cells treated with GSK1070916 shows dose-dependent inhibition of phosphorylation on serine 10 of Histone H3, a substrate specific for Aurora B. Moreover, GSK1070916 inhibits the proliferation of tumor cells with EC50 values of <10 nM in over 100 cell lines spanning a broad range of tumor types, with a median EC50 of 8 nM. Although GSK1070916 has potent activity against proliferating cells, a dramatic shift in potency is observed in primary, nondividing, normal human vein endothelial cells. Furthermore, GSK1070916-treated cells do not arrest in mitosis but instead fails to divide and become polyploid, ultimately leading to apoptosis. In another study, it is also reported high chromosome number associated with resistance to the inhibition of Aurora B and C suggests cells with a mechanism to bypass the high ploidy checkpoint are resistant to GSK1070916. in vivo: GSK1070916 (25, 50, or 100 mg/kg) shows dose-dependent inhibition of phosphorylation of an Aurora B–specific substrate in mice and consistent with its broad cellular activity, has antitumor effects in 10 human tumor xenograft models including breast, colon, lung, and two leukemia models.
Compound from plants keeps human cancer cells from multipying
Read more at Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
Weight control is an important concern of human beings, both for medical (pharmaceutical and/or nutraceutical) as well as non-therapeutic, e.g. cosmetic, reasons. More importantly, excessive accumulation of body fat (i.e. obesity (= adiposity), especially with excessive fat in the ventral region and surrounding the viscera) can be dangerous and has been linked to health problems such as type II diabetes, hypertension, heart disease, atherosclerosis (where more than two of the preceding disorders are present, the condition is often called “Metabolic Syndrome” or “syndrome X”), hyperlipidemia, coronary heart disease, stroke, breast and colon cancer, sleep apnoea, gallbladder disease, reproductive disorders such as polycystic ovarian syndrome, gastroesophageal reflux disease, increased incidence of complications of general anesthesia, fatty liver, gout or thromboembolism (see, e.g., Kopelman, Nature 404: 635-43 (2000)). Obesity reduces life-span and carries a serious risk of the co-morbidities listed above, as well disorders such as infections, varicose veins,
acanthosis nigricans, eczema, exercise intolerance, insulin resistance, hypertension hypercholesterolemia, cholelithiasis, orthopedic injury, and thromboembolic disease (Rissanen et al, Br. Med. J. 301 : 835-7 (1990)). Obesity is one of the main factors in the development of cardiovascular diseases. As a side effect the levels of cholesterol, blood pressure, blood sugar and uric acid in obese people are usually higher than those of persons of normal weight. The morbidity from coronary heart disease among the overweight people is increased as well. Among the people aged 40-50, mortality will rise about 1% when body weight increases by 0.5 kg and the death rate will increase 74% when body weight exceeds 25% of the standard. The prevalence of obesity in the United States has more than doubled since the turn of the last century (whole population) and more than tripled within the last 30 years among children aged from 6 to 11. This problem more and more becomes a disease risk also in Europe. In Germany, particularly many people have been found to suffer from overweight recently, already 25% of the young people, children and adolescents there are affected by obesity and related disorders. Furthermore, being overweight is considered by the majority of the Western population as unattractive.
Overweight and obesity result from an imbalance between the calories consumed and the calories used by the body. When the calories consumed exceed the calories burned, the body is in positive energy balance and over time weight gain will occur. The excess calories are stored in the fat cells. When the calories burned exceed the calories consumed, the body is in negative energy balance and over time weight loss will occur.
Determinants of obesity include social factors, psychological factors, genetic factors, developmental factors and decreased physical activity. Some components of a comprehensive weight loss programs include medical assessment, behavioural and dietary modification, nutrition education, mental and cognitive restructuring, increased physical activity, and long term follow-up.
An increasing interest by consumers in the maintenance or reduction of their body weight can be found. This leads to a demand for products useful for these purposes. Preferred are such food products which can conveniently be consumed as part of the daily diet, for example meal replacer products, such as meal replacer bars and beverages. These are usually designed for use as a single-serving food product to replace one or two meals a day.
An issue is that often a saturating effect is missed when such products are consumed, resulting in hunger feelings only a relatively short time after consummation or even in the lack of a saturation feeling already directly after consummation.
Summing up, there remains a need for new safe and effective compositions for promoting weight loss and/or loss of body fat in subjects such as humans. The problem to be solved by the present invention is therefore to find compositions or compounds useful in the treatment of obesity; and/or for improving the total cholesterol HDIJLDL ratio.
Phytochemistry provides a large pool of compounds and compositions to be looked at whether they are able to solve this problem.
The present invention provides methods and compositions useful in the control, treatment and prevention of obesity and obesity-related conditions, disorders, and diseases; and/or and/or for improving the total cholesterol HDL/LDL ratio.
Rosinski, G., et al., Endocrinological Frontiers in Phyiological Insect Ecology, Wroclow Technical University Press, Wroclow 1989, describe that certain tricyclic sequiterpene lactones, such as grossheimin and repin, showed inhibition of larval growth and antifeeding activity in Mealworm (Tenebrio σιοΐϊίοή. Grossheimin shows no anti-feeding but little decrease of absorption of digested food constituents and a little decrease in efficiency in digesting. Repin exhibit low effects at all. Both compounds show no effect on lipid levels in blood.
Shimoda, H., et al, Bioinorganic & Medicinal Chemistry Letters 13 (2003), 223-228, describe that methanolic extracts from Artichoke (Cynara sclolymus L.) with cynaropicrin, aguerin B and grossheimin as components and certain sesquiterpene glycosides suppress serum triglyceride elevation in olive oil-loaded mice. Some of these compounds exhibit a moderate short term (2 hours after olive oil administration) anti-hyperlipidemic activity presented as a lowering of the serum triglyceride (serum TG) concentrations, the long term (6 hours) show in the case of cynaropicrin and aguerine B an increase of the serum TG. Furthermore the authors present data of the gastric emptying (GE) of a methanolic ectract of artichoke. They determine a significantly inhibited GE. However, as shown below, this mechanism is not an explanation for the anti obesity effect shown in the present invention (see Example 1 ).
Fritzsche, J., et al., Eur. Food Res. Technol. 215, 149-157 (2002) describe the effect of certain isolated artichoke leaflet extract components with cholesterol lowering potential. Ahn, E.M-., et al, Arch Pharm. res. 29(1 1 ), 937-941 , 2006, shows ACAT inhibitory activity for two sesquiterpene lactones. KR 20040070985 also shows an effect of certain sesquiterpene lactone derivatives on cholesterol biosynthesis involved enzymes. Gebhard, R., Phytother. Res. 16, 368-372 (2002) and J. Pharmacol. Exp. Ther. 286(3), 1 122-1 128 (1998), shows
enforcement of cholesterol biosynthesis inhibition in HepG2 cells by artichoke extracts. WO 2007/006391 also claims reduction in cholesterol by certain Cynara scolymus variety extracts.
Other reported activities of tricyclic sesquiterpene lactones are antioxidant activity (European Food Research & Technology (2002), 215(2): 149-157), inhibitors of NF kb (Food Style 21 (2007), 1 1 (6): 54-56; JP 2006-206532), serum triglyceride increase-inhibitory effect (Kagaku Kogyo (2006), 57(10): 740-745), hypoglycaemic effect (J. Trad. Med. (2003), 20(2): 57-61), bitter taste (DE 2654184). Any beneficial effects are included in this invention by reference.
None of the documents suggest that a control and treatment of obesity and body fat in warmblooded animals might be possible.
Cynaropicrin, a tricyclic sesquiterpene lactone causes in vivo a strong weight loss. More surprisingly it was found that this effect is not correlated to a decrease in food intake. The weight balance is not affected by reduction of assimilation efficiency; the decrease of body fat and body weight is presumably caused by effects on energy metabolism. Surprisingly, it was found in addition that cynaropicrin also allows for improving the total cholesterol HDL7LDL ratio
Tricyclic sequiterpene lactones or known ingredients of plants of the subclass Asterides, especially from the family of Asteraceae, more specifically from species of the genera of the list consisting of Achilea, Acroptilon, Agranthus, Ainsliaea, Ajania, Amberboa, Andryala, Artemisia, Aster, Bisphopanthus, Brachylaena, Calea, Calycocorsus, Cartolepsis, Centaurea, Cheirolophus, Chrysanthemum, Cousinia, Crepis, Cynara, Eupatorium, Greenmaniella, Grossheimia, Hemistaptia, Ixeris, Jurinea, Lapsana, Lasiolaena, Liatris, Lychnophora, Macroclinidium, Mikania, Otanthus, Pleiotaxis, Prenanthes, Pseudostifftia, Ptilostemon,
Rhaponticum, Santolina, Saussurea, Serratula, Sonchus, Stevia, Taeckholmia, Tanacetum, Tricholepis, Vernonia, Volutarella, Zaluzania; even more specifically from species of the list consisting of Achillea clypeolata, Achillea collina, Acroptilon repens, Agrianthus pungens, Ainsliaea fragrans, Ajania fastigiata, Ajania fruticulosa, Amberboa lippi, Amberboa muricata, Amberboa ramose**, Amberboa tubuliflora and other Amberboa spp.*, Andryala integrifolia, Andryala pinnatifida, Artemisia absinthium, Artemisia cana, Artemisia douglasiana, Artemisia fastigiata, Artemisia franserioides, Artemisia montana, Artemisia sylvatica, Artemisia
parthenium, Tricholepis glaberrima** and other Tricholepsis spp. *, Vernonia arkansana, Vernonia nitidula, Vernonia noveboracensis, Vernonia profuga, Vernonia sublutea,
Volutarella divaricata, Zaiuzania resinosa; and can potentially be isolated from any part of the plants. Those genera and/or species marked with an asterisk (*) and especially those species marked with two asterisks (**) are especially preferred.
Appropriate plant material can be obtained from various sources, e.g. from:
Alfred Galke GmbH, Gittelde/Harz, Germany; Miiggenburg Pflanzliche Rohstoffe, Bad Bramstedt, Germany; Friedrich Nature Discovery, Euskirchen, Germany; VitaPlant AG, Uttwil, Switzerland; Amoros Nature SL, Hostalric, Spain.
(±)-Integrifolin
Banksia integrifolia
Coast Banksia
Family: Proteaceae
Banksia integrifolia is a tall shrub or small tree 6 – 16m tall. It is common in sandy coastal areas, but also grows in the forests of tablelands. The light grey bark is hard and rough.
Mature leaves 5 -10 cm long, are stiff, entire (untoothed), dull dark green above and hairy-white underneath. They are generally lanceolate. Younger leaves are irregularly toothed and shorter than the mature leaves. The species name ‘integrifolia’ means whole-leaved.
The pale yellow flower spikes of Banksia integrifolia range from 7-14cm long and 7cm wide. The bent styles emerge from individual flowers on the spike, straightening and spreading.
A short time after flowering, the seed pods protrude cleanly from the woody cone and open to shed black, papery, winged seeds.
Banksia integrifolia flowers from January to June.
SHENZHEN CHIPSCREEN BIOSCIENCES LTD. [CN/CN]; Research Institute of Tsinghua University, Suite C301, P.O. Box 28, High-Tech Industrial Park Nanshan District, Shenzhen, Guangdong 518057
As described for Example 2 according to the patent ZL03139760.3 obtained chidamide poor purity (about 95%).LC / MS analysis results shown in Figure 1, show that the product contains N- (2- amino-5-fluorophenyl) -4- (N- (3- pyridin-acryloyl group of 4.7% of the structure shown in formula II) aminomethyl) benzamide.1H NMR analysis of the results shown in Figure 2, show that the product contains 1.80% of tetrahydrofuran, far beyond the technical requirements for people with drug registration International Conference on Harmonization (ICH, International Conference of Harmonizition) provided 0.072% residual solvent limits.Therefore, the solid
Is NOT approved for the treatment of pancreatic cancer.
Chidamide drug administration and clinical milestone
November 2005: China declared IND
November 2006: eligible for Phase I clinical documents of approval
November 2006: completion of the International Patent Licensing, China entered the international fray original new drug development
May 2008: completed Phase I clinical, showing international mechanism similar drugs have the potential to become the best
February 2009: eligible lymphoma indications II / III of this document
March 2009: Start of the Phase II clinical trial for the NDA to ①CTCL goal of clinical trials and ②PTCL
March 2009: IND by the FDA application is eligible to start Phase I clinical in the United States
July 2009: eligible for non-small cell lung cancer, breast cancer and prostate cancer clinical documents of approval
December 2010: of PTCL by a conventional phase II directly into Phase II clinical trial registered drug trial center and by recognition
March 2011: combination chemotherapy for non-small cell lung cancer clinical trials enter phase Ib
September 2012: of PTCL indication test deadline
December 2012: of PTCL clinical summary will be held
January 2013: Chidamide declare China NDA
December 2014: the State Food and Drug Administration (CFDA) approved the listing
Chidamide overview, location and clinical significance
Chidamide (Chidamide, love spectrum sand ® / Epidaza®) Shenzhen microchip biotechnology limited liability company developed a new subtype selective histone having a chemical structure and is eligible for a global patent licensing deacetylase inhibitor, belong to the new mechanisms of epigenetic regulation new class of targeted anticancer drugs, has now completed with relapsed or refractory peripheral T-cell lymphoma clinical trial study registered indications, was in March 2013 to the SFDA reporting new drug certificate (NDA) and the marketing authorization (MAA). While a number of Chinese Cancer clinical trials undertaken Chidamide is also China’s first approved by the US FDA clinical studies in the United States of Chinese chemical original new drug trials in the United States Phase I has been completed. Chidamide has won the national “Eleventh Five-Year” 863 major projects (project number: 2006AA020603) and the national “Eleventh Five-Year”, “significant Drug Discovery” science and technology and other major projects funded project (project number: 2009ZX09401-003), was chosen the Ministry of Science and one of the “Eleventh five-Year” major national scientific and technological achievements.
Relapsed or refractory peripheral T-cell lymphoma (PTCL) is Chidamide first approvedclinical indications, PTCL belongs to the category of rare diseases, the lack of standard drug currently recommended clinical treatment, conventional chemotherapy response rate is low, recur, 5-year overall survival rate was about 25%. The world’s first PTCL treatment Folotyn (intravenous drug use) is eligible for FDA clearance to market in 2009, the second drugs Istodax (intravenous drug use) approved by the FDA in 2011. Add a new drug information for these drugs is very expensive, and were listed in China. Chidamide album clinical trial results showed that the primary endpoint of objective response rate was 28%, reaching the intended target research and development; sustained remission rate of 24% three months; drug safety was significantly better than the international similar drugs, and oral medication.
Chidamide is a completely independent intellectual property rights China originator of innovative medicines, has been multi-national patent. In China, for patients with relapsed or refractory PTCL to carry out effective drug treatment is urgent clinical need, Chidamide expected to bring new treatment options for patients with PTCL, prolong survival and improve quality of life of patients.
In China, for the effective treatment of patients with relapsed or refractory PTCL has undertaken urgent clinical need
Chidamide is a completely independent intellectual property rights China originator of innovative medicines
Chidamide (Chidamide) has been multi-national invention patents
In October 2006, the US HUYA biological microchip company formally signed the International Patent Chidamide licensing and international clinical cooperative development agreement; the United States in the ongoing Phase I clinical
Chidamide (Epidaza), a class I HDAC inhibitor, was discovered and developed by ChipScreen and approved by the CFDA in December 2014 for the treatment of recurrent of refractory peripheral T-cell lymphoma. Chidamide, also known as CS055 and HBI- 8000, is an orally bioavailable benzamide type inhibitor of HDAC isoenzymes class I , as well as class IIb 10, with potential antineoplastic activity. It selectively binds to and inhibits HDAC, leading to an increase in acetylation levels of histone protein H3.
Chidamide, the English called Chidamide, by the Shenzhen-core biotechnology limited liability company independent design and synthesis of a novel anti-cancer drugs with new chemical structures and global intellectual property, and its chemical name N- (2-amino-_4_ fluorophenyl) -4_ (N- (3- topiramate Li acryloyl) aminomethyl) benzamide, its chemical structure of the structural formula I
The patent ZL03139760.3 and said US7,244,751, Chidamide have histone deacetylase inhibitory activity can be used to treat the differentiation and proliferation-related diseases such as cancer and psoriasis, especially for leukemia and solid tumors with excellent results.
Patent No. ZL03139760.3 and US7,244,751 discloses a method for preparing chidamide, but did not specify whether the resulting product is a crystalline material, nor did the presence or absence of the compound polymorphism.In the above patent, the activity of the compound for evaluation is not conducted in a solid state and, therefore, does not disclose any description about characteristics of the crystal.
Chipscreen BioSciences announced that the CFDA had approved chidamide for the treatment of relapsed or refractory peripheral T-cell lymphoma (PTCL) in December 2014. The drug and Hengrui’s apatinib were the only two NCEs launched by domestic drug makers last year.
Chidamide (CS055/HBI-8000) is a HDAC1/2/3/10 inhibitor derived from entinostat (MS-27-275)[1] which was first discoved by Mitsui Pharmaceuticals in 1999. Chipscreen holds worldwide IP rights to chidamide (patents: WO2004071400, WO2014082354).
Syndax Pharmaceuticals (NASDAQ: SNDX) is testing entinostat in breast cancer and NSCLC in pivotal trials. The FDA granted Breakthrough Therapy Designation to entinostat for advanced breast cancer in 2013. Eddingpharm in-licensed China rights to entinostat from Syndax in September 2013.
Chipscreen disclosed positive results from Phase II study of chidamide in relapsed or refractory PTCL at 2013 ASCO Annual Meeting[2]. Out of 79 evaluable patients in the trial, 23 patients (29.1%) had confirmed responses (8 CR, 3 CRu, and 12 PR). The most common grade 3/4 AEs were thrombocytopenia (24%), leucocytopenia (13%), neutropenia(10%).
The FDA has approved three HDAC inhibitors, known as Zolinza (vorinostat), Istodax (romidepsin) and Beleodaq (belinostat), for the treatment of PTCL. Celgene priced Istodax at $12000-18000/month and reported annual sales of $54 million in 2013. The efficacy and safety profile of chidamide compares favorably with romidepsin.
Although a dozen of companies are developing generic vorinostat and romidepsin, no chemical 3.1 NDA has been submitted to the CFDA so far. Chipscreen will be the only domestic maker of HDAC inhibitor in the coming two years. Moreover, the company is testing chidamide in NSCLC and breast cancer in early clinical studies.
CLIP
Chiamide synthesis: US7244751B2
Procedure:
Step a: To a suspension of 0.33 g (2.01 mmol) of N,N’-carbonyldiimidazole in tetrahydrofunan (10 ml) is added drop-wise a solution of 0.30 g (2.01 mmol) of 3-pyridineacrylic acid at 0 °C. Then, the mixture is stirred at room temperature for 3 hours and added drop-wise to a separately prepared 2.0 ml (2.00 mmol) of 1N aqueous sodium hydroxide solution including 0.30 g (2.00 mmol) of 4-aminomethylbenzoic acid, followed by stirring at room temperature for 8 hours. The reaction mixture is evaporated under vacuum. To the residue is added a saturated solution of sodium chloride (2 ml), then the mixture is neutralized with concentrated hydrochloric acid to pH 5. The deposited white solid is collected by filtration, washed with ice-water, and then dried to give 4-[N-(Pyridin-3-ylacryloyl)aminomethyl]benzoic acid (0.46 g, 82%). HRMS calcd for C16H14N2O3: 282.2988. Found: 282.2990. MA calcd for: C16H14N2O3: C, 68.07%; H, 5.00%; N, 9.92%. Found: C, 68.21%; H, 5.03%; N, 9.90%.
Step b: To a suspension of 0.29 g (1.78 mmol) of N,N’-carbonyldiimidazole in tetrahydrofunan (15 ml) is added 0.50 g (1.78 mmol) of 4-[N-(Pyridin-3-ylacryloyl)aminomethyl]benzoic acid, followed by stirring at 45 °C. for 1 hour. After cooling, the reaction mixture is added to a separately prepared tetrahydrofiman (10 ml) solution including 0.28 g (2.22 mmol) of 4-fluoro-1,2-phenylenediamine and 0.20 g (1.78 mmol) of trifluoroacetic acid at room temperature. After reaction at room temperature for 24 hours, the deposited white solid is collected by filtration, washed with tetrahydrofunan, and then dried to give N-(2-amino-4-fluorophenyl)-4-[N-(Pyridin-3-ylacryloyl)aminomethyl]benzamide (0.40 g, 57%). 1H NMR (300 MHz, DMSO-d6): dppm: 4.49 (2H, d), 4.84 (2H, br.s), 6.60 (1H, t), 6.80 (2H, m),696 (1H, t), 7.18 (1H, d), 7.42 (2H, d), 7.52 (1H, d), 7.95 (2H, d), 8.02 (1H, d), 8.56 (1H, d), 8.72 (1H, br. t), 8.78 (1H, s), 9.60 (1H, br.s). IR (KBr) cm1: 3310, 1655, 1631, 1524, 1305, 750. HRMS calcd for C22H19N4O2F: 390.4170. Found: 390.4172. MA calcd for C22H19N4O2F: C, 67.68%; H, 4.40%; N, 14.35%. Found: C, 67.52%; H, 4.38%; N, 14.42%.
Preparation of 4-[N-(Pyridin-3-ylacryloyl)aminomethyl]benzoic acid
To a suspension of 0.33 g (2.01 mmol) of N,N′-carbonyldiimidazole in tetrahydrofunan (10 ml) is added drop-wise a solution of 0.30 g (2.01 mmol) of 3-pyridineacrylic acid at 0° C. Then, the mixture is stirred at room temperature for 3 hours and added drop-wise to a separately prepared 2.0 ml (2.00 mmol) of 1N aqueous sodium hydroxide solution including 0.30 g (2.00 mmol) of 4-aminomethylbenzoic acid, followed by stirring at room temperature for 8 hours. The reaction mixture is evaporated under vacuum. To the residue is added a saturated solution of sodium chloride (2 ml), then the mixture is neutralized with concentrated hydrochloric acid to pH 5. The deposited white solid is collected by filtration, washed with ice-water, and then dried to give the title compound (0.46 g, 82%). HRMS calcd for C16H14N2O3: 282.2988. Found: 282.2990. MA calcd for: C16H14N2O3: C, 68.07%; H, 5.00%; N, 9.92%. Found: C, 68.21%; H, 5.03%; N, 9.90%.EXAMPLE 2
Preparation of N-(2-amino-4-fluorophenyl)-4-[N-(Pyridn-3-ylacryloyl)aminomethyl]benzamide
To a suspension of 0.29 g (1.78 mmol) of N,N′-carbonyldiimidazole in tetrahydrofunan (15 ml) is added 0.50 g (1.78 mmol) of 4-[N-(Pyridn-3-ylacryloyl)aminomethyl]benzoic acid, followed by stirring at 45° C. for 1 hour. After cooling, the reaction mixture is added to a separately prepared tetrahydrofiman (10 ml) solution including 0.28 g (2.22 mmol) of 4-fluoro-1,2-phenylenediamine and 0.20 g (1.78 mmol) of trifluoroacetic acid at room temperature. After reaction at room temperature for 24 hours, the deposited white solid is collected by filtration, washed with tetrahydrofunan, and then dried to give the title compound (0.40 g, 57%). 1H NMR (300 MHz, DMSO-d6): δppm: 4.49 (2H, d), 4.84 (2H, br.s), 6.60 (1H, t), 6.80 (2H, m),696 (1H, t), 7.18 (1H, d), 7.42 (2H, d), 7.52 (1H, d), 7.95 (2H, d), 8.02 (1H, d), 8.56 (1H, d), 8.72 (1H, br. t), 8.78 (1H, s), 9.60 (1H, br.s). IR (KBr) cm1: 3310, 1655, 1631, 1524, 1305, 750. HRMS calcd for C22H19N4O2F: 390.4170. Found: 390.4172. MA calcd for C22H19N4O2F: C, 67.68%; H, 4.40%; N, 14.35%. Found: C, 67.52%; H, 4.38%; N, 14.42%.EXAMPLE 3
Preparation of 4-[N-cinnamoylaminomethyl]benzoic acid
To a suspension of 0.33 g (2.01 mmol) of N,N′-carbonyldiimidazole in tetrahydrofunan (10 ml) is added drop-wise a solution of 0.30 g (2.01 mmol) of cinnamic acid at 0° C. Then, the mixture is stirred at room temperature for 3 hours and added drop-wise to a separately prepared 2.0 ml (2.00 mmol) of 1N aqueous sodium hydroxide solution including 0.30 g (2.00 mmol) of 4-aminomethylbenzoic acid, followed by stirring at room temperature for 8 hours. The reaction mixture is evaporated under vacuum. To the residue is added a saturated solution of sodium chloride (2 ml), then the mixture is neutralized with concentrated hydrochloric acid to pH 7. The deposited white solid is collected by filtration, washed with ice-water, and then dried to give the title compound (0.51 g, 91%). HRMS calcd for C17H15NO3: 281.3242. Found: 281.3240. MA calcd for C17H15NO3: C, 72.58%; H, 5.38%; N, 4.98. Found: C, 72.42%; H, 5.37%; N, 4.98%.
EXAMPLE 4
Preparation of N-(2-amino-4-fluorophenyl)-4-[N-cinnamoylaminomethyl]benzamide
To a suspension of 0.29 g (1.78 mmol) of N,N′-carbonyldiimidazole in tetrahydrofunan (15 ml) is added 0.50 g (1.78 mmol) of 4-[N-cinnamoylaminomethyl]benzoic acid, followed by stirring at 45° C. for 1 hour. After cooling, the reaction mixture is added to a separately prepared tetrahydrofunan (10 ml) solution including 0.28 g (2.22 mmol) of 4-fluoro-1,2-phenylenediamine and 0.20 g (1.78 mmol) of trifluoroacetic acid at room temperature. After reaction at room temperature for 16 hours, the deposited white solid is collected by filtration, washed with tetrahydrofunan, and then dried to give the title compound (0.45 g, 64%). 1H NMR (300 MHz, DMSO-d6): δppm: 4.42 (2H, d), 4.92 (2H, br.s), 6.62 (1H, t), 6.78 (2H, m), 7.01 (1H, t), 7.32 (5H, m), 7.54 (5H, m), 8.76 (1H, br.t), 9.58 (1H, br.s). IR (KBr) cm−1: 3306, 1618, 1517, 1308, 745. HRMS calcd for C23H20N3O2F: 389.4292. Found: 389.4294. MA calcd for C23H20N3O2F: C, 70.94%; H, 5.18%; N, 10.79%. Found: C, 70.72%; H, 5.18%; N, 10.88%.
Chidamide (Epidaza), a class I HDAC inhibitor, was discovered and developed by ChipScreen and approved by the CFDA in December 2014 for the treatment of recurrent of refractory peripheral T-cell lymphoma. Chidamide, also known as CS055 and HBI- 8000, is an orally bioavailable benzamide type inhibitor of HDAC isoenzymes class I 1–3, as well as class IIb 10, with potential antineoplastic activity. It selectively binds to and inhibits HDAC, leading to an increase in acetylation levels of histone protein H3.74
This agent also inhibits the expression of signaling kinases in the PI3K/ Akt and MAPK/Ras pathways and may result in cell cycle arrest and the induction of tumor cell apoptosis.75
Currently, phases I and II clinical trials are underway for the treatment of non-small cell lung cancer and for the treatment of breast cancer, respectively.76 The scalable synthetic approach to chidamide very closely follows the discovery route,77–79 and is described in Scheme 10. The sequence began with the condensation of commercial nicotinaldehyde (52) and malonic acid (53) in a mixture of pyridine and piperidine. Next, activation of acid 54 with N,N0-carbonyldiimidazole (CDI) and subsequent reaction with 4-aminomethyl benzoic acid (55) under basic conditions afforded amide 56 in 82% yield.
Finally, activation of 56 with CDI prior to treatment with 4-fluorobenzene- 1,2-diamine (57) and subsequent treatment with TFA and THF yielded chidamide (VIII) in 38% overall yield from 52. However, no publication reported that mono-N-Boc-protected bis-aniline was used to approach Chidamide.
74. Ning, Z. Q.; Li, Z. B.; Newman, M. J.; Shan, S.; Wang, X. H.; Pan, D. S.; Zhang, J.;
Dong, M.; Du, X.; Lu, X. P. Cancer Chemother. Pharmacol. 2012, 69, 901.
75. Liu, L.; Chen, B.; Qin, S.; Li, S.; He, X.; Qiu, S.; Zhao, W.; Zhao, H. Biochem.
Biophys. Res. Commun. 2010, 392, 190.
76. Gong, K.; Xie, J.; Yi, H.; Li, W. Bio. Chem. J. 2012, 443, 735.
77. Lu, X. P.; Li, Z. B.; Xie, A. H.; Shi, L. M.; Li, B. Y.; Ning, Z. Q.; Shan, S.; Deng, T.;
Hu, W. M. US Patent 2004224991A1, 2004.
78. Lu, X. P.; Li, Z. B.; Xie, A. H.; Shi, L. M.; Li, B. Y.; Ning, Z. Q.; Shan, S.; Deng, T.;
Hu, W. M. CN Patent 1513839A, 2003.
79. Yin, Z. H.; Wu, Z. W.; Lan, Y. K.; Liao, C. Z.; Shan, S.; Li, Z. L.; Ning, Z. Q.; Lu, X.
P.; Li, Z. B. Chin. J. New Drugs 2004, 13, 536.
Example 2. Preparation of
N-(2-amino-5-fluorophenyl)-4-[N-(Pyridn-3-ylacryloyl)aminomethyl]benzamide
To a suspension of 0.29 g (1.78 mmol) of N, N’-carbonyldiimidazole in tetrahydrofunan (15 ml) is added 0.50 g (1.78 mmol) of 4-[N-(Pyridn-3-ylacryloyl)aminomethyl]benzoic acid, followed by stirring at 45°C for 1 hour. After cooling, the reaction mixture is added to a separately prepared tetrahydrofunan (10 ml) solution including 0.28 g (2.22 mmol) of 4-fluoro-1,2-phenylenediamine and 0.20 g (1.78 mmol) of trifluoroacetic acid at room temperature. After reaction at room temperature for 24 hours, the deposited white solid is collected by filtration, washed with tetrahydrofunan, and then dried to give the title compound (0.40 g, 57%). 1H NMR (300 MHz, DMSO-d6): δppm: 4.49 (2H, d), 4.84 (2H, br.s), 6.60 (IH, t), 6.80 (2H, m), 6.96 (IH, t), 7.18 (IH, d), 7.42 (2H, d), 7.52 (IH, d), 7.95 (2H, d), 8.02 (IH, d), 8.56 (IH, d), 8.72 (IH, br. t), 8.78 (IH, s), 9.60 (IH, br.s). IR (KBr) cm“1: 3310, 1655, 1631, 1524, 1305, 750. HRMS calcd for C22Hι9N4O2F: 390.4170. Found: 390.4172. MA calcd for C22Hι9N4O2F: C, 67.68%; H, 4.40%; N, 14.35. Found: C, 67.52%; H, 4.38%; N, 14.42%.
Photo taken on May 22, 2015 shows a box of Chidamide in Shenzhen, south China’s Guangdong Province. Chidamide is the world’s first oral HDAC inhibitor …
A New Cancer Drug, Made in China
After 14 years, Shenzhen biotech’s medicine is one of the few locally developed from start to finish
Xian-Ping Lu left his research job at a drug maker in the U.S. to co-found a biotech company in his native China.PHOTO: SHENZHEN CHIPSCREEN BIOSCIENCES
HONG KONG— Xian-Ping Lu left his job as director of research at drug maker Galderma R&D in Princeton, N.J., to co-found a biotech company to develop new medicines in his native China.
It took more than 14 years but the bet could be paying off. In February, Shenzhen Chipscreen Biosciences’ first therapy, a medication for a rare type of lymph-node cancer, hit the market in China.
The willingness of veterans like Dr. Lu and others to leave multinational drug companies for Chinese startups reflects a growing optimism in the industry here. The goal, encouraged by the government, is to move the Chinese drug industry beyond generic medicines and drugs based on ones developed in the West.
Chipscreen’s drug, called chidamide, or Epidaza, was developed from start to finish in China. The medicine is the first of its kind approved for sale in China, and just the fourth in a new class globally. Dr. Lu estimates the research cost of chidamide was about $70 million, or about one-tenth what it would have cost to develop in the U.S.
“They are a good example of the potential for innovation in China,” said Angus Cole, director at Monitor Deloitte and pharmaceuticals and biotechnology lead in China.
China’s spending on pharmaceuticals is expected to top $107 billion in 2015, up from $26 billion in 2007, according to Deloitte China. It will become the world’s second-largest drug market, after the U.S., by 2020, according to an analysis published last year in the Journal of Pharmaceutical Policy and Practice.
China has on-the-ground infrastructure labs, a critical mass of leading scientists and interested investors, according to Franck Le Deu, head of consultancy McKinsey & Co.’s pharmaceuticals and medical-products practice in China. “There’re all the elements for the recipe for potential in China,” he said.
But there are obstacles to an industry where companies want big payoffs for a decade or more of work and tremendous costs it takes to develop a drug.
While the protection of intellectual property has improved, China’s cumbersome rules for drug approval and a government effort to cut health-care costs, particularly spending on drugs, could hurt the Chinese drug companies’ efforts, said Mr. Cole of Deloitte.
“Will you start to see success? Of course you will,” said Mr. Cole. However, “I’ve yet to see convincing or compelling evidence that it’s imminent.”
To date, many of the Chinese companies that are flourishing in the life sciences are contract research organizations that help carry out clinical trials, as well as providers of related services.
Some companies, like Shanghai-based Hua Medicine, are buying the rights to develop new compounds in China from multinational drug companies, what some experts consider more akin to an intermediate step to innovation.
Late last year, Hua Medicine completed an early-stage human clinical trial of a diabetes drug in China and in March filed an application to the Food and Drug Administration to develop it in the U.S. as well. The company has raised $45 million in venture funding to date.
Li Chen, who left an 18-year career at Roche Holding AG as head of research and development in China to help start Hua Medicine, said the company’s goal is to “create a game-changer of drug discovery.”
At Chipscreen Biosciences, Dr. Lu and his co-founders set up the company in 2001 in Shenzhen, a city that was quickly growing into a technology and research hub, just over the border from Hong Kong. They created a lab of 10 scientists to use a new analytic technique known as “chemical genomics” to examine the relationships between molecular structures of the existing and failed drugs, how they act on different targets in the body and what genes were being activated or repressed. Now they have more than 60 scientists.
By better predicting how chemicals would act on the body before entering human testing, they hoped they would be more likely get a drug to market.
“How can a small company compete with a multinational?” said Dr. Lu. “The only thing we can compete with is the scientific brain.”
The biggest challenges for the company have been financing and the Chinese regulatory system, said Dr. Lu. The company has raised a total of 300 million yuan ($48 million) over five rounds of venture funding, said Dr. Lu. Chipscreen also receives grant money from the Chinese government.
The company filed its application for approval of chidamide to the Chinese Food and Drug Administration, or CFDA, in early 2013. It had to wait nearly two years for approval, receiving the OK only in December.
Chidamide now is on the market in China for 26,500 yuan ($4,275) a month, a price far lower than patients in the U.S. pay for some of the newest cancer medicines but much more than the typical Chinese patient pays for drugs. Dr. Lu said the price reflects a balance between affordability for patients and return for shareholders. Some investors wanted to price the drug higher.
PAPER
Discovery of an orally active subtype-selective HDAC inhibitor, chidamide, as an epigenetic modulator for cancer treatment
Tumorigenesis is maintained through a complex interplay of multiple cellular biological processes and is regulated to some extent by epigenetic control of gene expression. Targeting one signaling pathway or biological function in cancer treatment often results in compensatory modulation of others, such as off-target drivers of cell survival. As a result, overall survival of cancer patients is still far from satisfactory. Epigenetic-modulating agents can concurrently target multiple aberrant or compensatory signaling pathways found in cancer cells. However, existing epigenetic-modulating agents in cancer treatment have not yet fully translated into survival benefits beyond hematological tumors. In this article, we present a historical rationale for use of chidamide (CS055/Epidaza), an orally active and subtype-selective histone deacetylase (HDAC) inhibitor of the benzamide chemical class. This compound was discovered and successfully developed as mono-therapy for relapsed and refractory peripheral T cell lymphoma (PTCL) in China. We discuss the evidence supporting chidamide as a durable epigenetic modulator that allows cellular reprogramming with little cytotoxicity in cancer treatments.
CLIPS
Chinese scientists develop world’s 1st oral HDAC inhibitor
Lu Xianping works in a lab at Shenzhen Chipscreen Biosciences Ltd. in Shenzhen, south China’s Guangdong Province, May 20, 2015. Lu Xianping, together with other four returned overseas scientists, spent 14 years to develop Chidamide, the world’s first oral HDAC inhibitor, which was given regulatory approval in January. (Xinhua/Mao Siqian)
GNT Biotech and Medicals Corporation Licenses Novel Cancer Molecule from Shenzhen Chipscreen Biosciences Ltd.
PR Newswire
SHENZHEN, China, Oct. 10, 2013
SHENZHEN, China, Oct. 10, 2013 /PRNewswire/ — GNT Biotech and Medicals Corporation announces the grant of an exclusive license from Shenzhen Chipscreen Biosciences Ltd.for the development and commercialization of Chidamide in Taiwan. Chidamide, an oral, selective histone deacetylase (HDAC) inhibitor, is currently being evaluated in Phase II trials by Chipscreen Biosciences in Peripheral T-Cell Lymphoma (PTCL), Cutaneous T-Cell Lymphoma (CTCL) and Non-Small Cell Lung Cancer patients (NSCLC). GNTbm will develop and commercialize Chidamide primarily in PTCL, NSCLC and will also retain the rights to develop and commercialize Chidamide in other oncology indications in Taiwan.
About Chidamide
Chidamide is a selective HDAC inhibitor against subtype 1, 2, 3 and 10, and being studied in multiple clinical trials as a single agent or in combination with chemotherapeutic agents for the treatment of various hematological and solid cancers. Its anticancer effects are thought to be mediated through epigenetic modulation via multiple mechanisms of action, including the inhibition of cell proliferation and induction of apoptosis in blood derived cells, inhibition of epithelial to mesenchymal transition (EMT, a process that is highly relevant to tumor cell metastasis and drug resistance), induction of tumor specific antigen and antigen-specific T cell cytotoxicity, enhancement of NK cell anti-tumor activity, induction of cancer stem cell differentiation, and resensitization of tumor cells that have become resistant to anticancer agents such as platinums, taxanes and topoisomerase II inhibitors. Chidamide has demonstrated clinical efficacy in pivotal phase II trials on Cutaneous T-Cell Lymphoma (CTCL) and Peripheral T-Cell Lymphoma (PTCL) conducted in China, and is currently undergoing phase II trial in NSCLC together with first line PC therapeutic treatment. Due to its superior pharmacokinetic properties and selectivity, Chidamide may offer better clinical profile over the other HDAC inhibitors currently under development or being marketed.
About GNTbm
GNTbm is a subsidiary of GNT Inc, a Taiwanese company focused on the manufacture of nano-scale metallic particles for food and medical purposes. Founded in 1992 by a team of electronic professionals, GNT has successfully developed the innovative technology of physical metal miniaturization based on the patent of MBE (Molecular Beam Epitaxy). Further information about GNT Inc is available at www.gnt.com.tw.
GNTbm was established in August 2013, and housed in the Nankang Biotech Incubation Center, (NBIC), in Nankang, Taipei. Lead by Dr. Chia-Nan Chenalong with an experienced team of scientists, GNTbm will explore development and commercialization of novel drug delivery systems, Innovative biomedical and diagnostic tools based on gold nanoparticles.
About Shenzhen Chipscreen Biosciences Ltd.
Chipscreen is a leading integrated biotech company in China specialized in discovery and development of novel small molecule pharmaceuticals. The company has utilized its proprietary chemical genomics-based discovery platform to successfully develop a portfolio of clinical and preclinical stage programs in a number of therapeutic areas. Chipscreen’s business strategy is to generate differentiated drug candidates across multiple therapeutic areas. Drug candidates are either developed by Chipscreen or co-developed and commercialized in a partnership at the research, preclinical and clinical stages. The company was established as Sino-foreign joint venture in 2001. Further details about Chipscreen Bioscience is available atwww.chipscreen.com.
Qiao, Z (2013-04-26). “Chidamide, a novel histone deacetylase inhibitor, synergistically enhances gemcitabine cytotoxicity in pancreatic cancer cells.”. Biochem Biophys Res Commun.434 (1): 95–101. doi:10.1016/j.bbrc.2013.03.059. PMID23541946.
References:
1. Ning, Z. Q.; et. al. Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother Pharmacol2012, 69(4), 901-909. (activity)
2. Gong, K.; et. al. CS055 (Chidamide/HBI-8000), a novel histone deacetylase inhibitor, induces G1 arrest, ROS-dependent apoptosis and differentiation in human leukaemia cells. Biochem J 2012, 443(3), 735-746. (activity)
3. Hu, W.; et. al. N-(2-amino-5-fluorophenyl)-4-[N-(Pyridin-3-ylacryloyl) aminomethyl ]benzamide or other derivatives for treating cancer and psoriasis. US7244751B2
4. Lu, X.; et. al. Crystal form of chidamide, preparation method and use thereof. WO2014082354A1
5. Yin, Z.-H.; et. al. Synthesis of chidamide,a new histone deacetylase (HDAC) inhibitor. Chin J New Drugs 2004, 13(6), 536-538. (starts with basic raw materials)
Zhongguo Xinyao Zazhi (2004), 13(6), 536-538.
/////////Chidamide, Epidaza, CS055, HBI-8000, orally active subtype-selective HDAC inhibitor, epigenetic modulator, cancer treatment, CFDA, CHINA, CANCER
A MEK1/Raf inhibitor potentially for the treatment of solid tumors and multiple myeloma.
RO-5126766; RG-7304; CH-5126766; CKI-27; R-7304
CAS No. 946128-88-7
Although melanoma is the most aggressive skin cancer, recent advances in BRAF and/or MEK inhibitors against BRAF-mutated melanoma have improved survival rates. Despite these advances, a treatment strategy targeting NRAS-mutated melanoma has not yet been elucidated. We discovered CH5126766/RO5126766 as a potent and selective dual RAF/MEK inhibitor currently under early clinical trials. We examined the activity of CH5126766/RO5126766 in a panel of malignant tumor cell lines including melanoma with a BRAF or NRAS mutation. Eight cell lines including melanoma were assessed for their sensitivity to the BRAF, MEK, or RAF/MEK inhibitor using in vitro growth assays. CH5126766/RO5126766 induced G1 cell cycle arrest in two melanoma cell lines with the BRAF V600E or NRAS mutation. In these cells, the G1 cell cycle arrest was accompanied by up-regulation of the cyclin-dependent kinase inhibitor p27 and down-regulation of cyclinD1. CH5126766/RO5126766 was more effective at reducing colony formation than a MEK inhibitor in NRAS- or KRAS-mutated cells. In the RAS-mutated cells, CH5126766/RO5126766 suppressed the MEK reactivation caused by a MEK inhibitor. In addition, CH5126766/RO5126766 suppressed the tumor growth in SK-MEL-2 xenograft model
A method for producing a coumarin derivative of general formula (VII) is disclosed in Patent document 1 or 2. Patent document 1 or 2 discloses a method represented by the scheme below [In the scheme, DMF represents N,N-dimethylformamide, TBS represents a tert-butyldimethylsilyl group, dba represents dibenzylideneacetone, and BINAP represents 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl. Also, the numerical values (%) and “quant.” given below some structural formulas indicate the yields of the respective compounds], for example (see the manufacturing example for “compound 1j-2-16-2K” in Patent document 1 or 2).
Methylamine (158 µL, 317 µmol) and DMAP (38.7 mg, 317 µmol) were added at -78 °C to a solution of sulfuryl chloride (28 µL, 340 µmol) in dichloromethane (2 mL), and the mixture was then stirred at room temperature for 2 hours to yield the corresponding sulfamoyl chloride. 3-(2-Amino-3-fluoropyridin-4-ylmethyl)-7-(pyrimidin-2-yloxy)-4-methyl-2-oxo-2H-1-benzopyran (compound 1h-2-16) (60 mg, 159 µmol), pyridine (65 µL, 795 µmol) and dichloromethane (2 mL) were added to the reaction solution, and the mixture was stirred at room temperature for 4 hours. After addition of water, the organic layer was extracted with dichloromethane. After washing with sodium hydrogen carbonate solution and saturated saline, the organic layer was dried over anhydrous magnesium sulfate, and the solvent was distilled away under reduced pressure. The resultant residue was purified by silica gel column chromatography to yield the title compound (32 mg, 43%).
3-(2-(N-Methylsulfamoyl)amino-3-fluoropyridin-4-ylmethyl)-4-methyl-7-(pyrimidin-2-yloxy)-2-oxo-2H-1-benzopyran sodium salt
The title compound was synthesized under the same conditions as in the manufacturing example for compound 1j-1-5-1Na, except that compound 1j-2-16-2 was used instead of compound 1j-1-5-1.
3-(2-(N-Methylsulfamoyl)amino-3-fluoropyridin-4-ylmethyl)-4-methyl-7-(pyrimidin-2-yloxy)-2-oxo-2H-1-benzopyran potassium salt
The title compound was synthesized under the same conditions as in the manufacturing example for compound 1j-1-5-1Na, except that compound 1j-2-16-2 was used instead of compound 1j-1-5-1, and that KOH was used instead of NaOH.
† Research Division, Chugai Pharmaceutical Co., Ltd., 200 Kajiwara, Kamakura, Kanagawa 247-8530, Japan
‡ Research Division, Chugai Pharmaceutical Co., Ltd., 1-135 Komakado, Gotemba, Shizuoka 412-8513, Japan
ACS Med. Chem. Lett., 2014, 5 (4), pp 309–314
DOI: 10.1021/ml400379x
Publication Date (Web): January 22, 2014
Substituting a carbon atom with a nitrogen atom (nitrogen substitution) on an aromatic ring in our leads 11a and 13g by applying nitrogen scanning afforded a set of compounds that improved not only the solubility but also the metabolic stability. The impact after nitrogen substitution on interactions between a derivative and its on- and off-target proteins (Raf/MEK, CYPs, and hERG channel) was also detected, most of them contributing to weaker interactions. After identifying the positions that kept inhibitory activity on HCT116 cell growth and Raf/MEK, compound 1(CH5126766/RO5126766) was selected as a clinical compound. A phase I clinical trial is ongoing for solid cancers.
Step 5 Synthesis of 4-methyl-3-(3-fluoro-2-aminopyridin-4-ylmethyl)-7-(pyrimidin-2-yloxy)-2-oxo-2H-1-benzopyran
Under a nitrogen atmosphere, potassium carbonate (2.3 g, 17 mmol) was added to a solution of the solid product of step 4 (3.0 g) and 2-bromopyrimidine (1.6 g, 9.8 mmol) in DMF (48 mL), and the mixture was stirred at 115° C. for 2.5 hours. The reaction mixture was cooled to 28° C., water (48 mL) was added dropwise over a period of 5 minutes at that temperature, and after cooling to 0° C., the mixture was stirred for 2 hours. The precipitated crystals were collected by filtration, washed with water (24 mL) and acetonitrile (24 mL) in that order, and dried under reduced pressure to obtain crude crystals (2.3 g). DMF (65 mL) was added to the crude crystals (2.3 g), and after heating to 60° C. and confirming the dissolution, the mixture was cooled to 25° C. Water (65 mL) was added at 25° C., and the mixture was further cooled to 0° C. and stirred for 4 hours. The precipitated crystals were collected by filtration, washed with water (22 mL) and acetonitrile (22 mL) in that order, and dried under reduced pressure to obtain the title compound (2.1 g). The title compound is a compound disclosed in WO 2007/091736.
Yield (overall yield from the 2-acetylamino-5-chloro-3-fluoropyridine used in step 2): 27%
The title compound was synthesized under the same conditions as in the manufacturing example for compound 1h-2-4 (synthesis scheme 2), except that compound 5d-0-16 was used instead of compound 4a-0-4.
The title compound was synthesized under the same conditions as in the manufacturing example for compound 1j-1-5-2, except that compound 1h-2-4 was used instead of compound 1h-1-5.
One of the CH3 peaks was overlapped with the DMSO peak.
ESI (LC/MS positive mode) m/z: 471 (M+H).
Compound 1j-2-4-2Na:
3-{2-Fluoro-3-(methylaminosulfonyl)aminobenzyl}-4-methyl-7-(pyrimidin-2-yloxy)-2-oxo-2H-1-benzopyran sodium salt
The title compound was synthesized under the same conditions as in the manufacturing example for compound 1j-1-5-1Na, except that compound 1j-2-4-2 was used instead of compound 1j-1-5-1.
3-{2-Fluoro-3-(methylaminosulfonyl)aminobenzyl}-4-methyl-7-(pyrimidin-2-yl-oxy)-2-oxo-2H-1-benzopyran potassium salt
The title compound was synthesized under the same conditions as in the manufacturing example for compound 1j-1-5-1Na, except that compound 1j-2-4-2 was used instead of compound 1j-1-5-1, and that KOH was used instead of NaOH.
Method for producing a coumarin derivative of formula (VII) are described in Patent Documents 1 and 2.Patent Documents 1 and 2, for example, in the following scheme [scheme, DMF is N, represents a N- dimethylformamide, TBS represents a tert- butyldimethylsilyl group, dba represents dibenzylideneacetone, BINAP is 2, I represents a 2′-bis (diphenylphosphino) -1,1′-binaphthyl.Further, numerical values given under the formula (%) or “quant.” Indicates the yield of the compound.Methods have been described that are shown in (see Preparation of “Compound 1j-2-16-2K” in Patent Documents 1 and 2).
WO2007 / 091736WO2009 / 014100
While coumarin derivatives of the general formula (VII) can be prepared by the methods described in Patent Documents 1 and 2, in the method described in Patent Documents 1 and 2, after the formylation reaction and a reduction reaction, and unintended Reaction To suppress, it is necessary to perform the introduction and removal steps of the protecting group for hydroxy group.Also, during the formylation reaction, from the viewpoint of cryogenic conditions of the reaction control (eg, -95 ℃ ~ -65 ℃) is required.Furthermore, the alkylation reaction (the seventh step in the above scheme), it is preferred that an excess amount of use of ethyl acetoacetate in terms of efficient synthesis, in which case, requires complicated operation of removing residual reagents become.
[Example 1] Step 1: Synthesis of 2-acetylamino-5-chloro-3-fluoropyridine:
Under a nitrogen atmosphere, acetamide (94.8g, 1.61mol) in DMF with (200mL) and THF (830mL) was added and heated to 50 ℃.The resulting solution was a THF solution of 40wt% sodium hexamethyldisilazide (629g, 1.37mol) was added dropwise and stirred at the same temperature for 2 hours.5-chloro-2,3-difluoro pyridine (100.0g, 0.67mol) After adding, THF and (20mL), and the mixture was stirred at the same temperature for 3 hours.After cooling to 0 ℃, it is added to 2.8M HCl (500mL) to the reaction mixture, and the organic layer was separated and the temperature was raised to room temperature.The organic layer was washed with 20wt% sodium chloride solution (500mL), and evaporated under reduced pressure.The residue in THF (500mL) was added, and the residue was dissolved by heating at 70 ℃.After confirming the solid precipitated by cooling to room temperature, n- heptane (1500mL) was added and further cooled to 0 ℃, followed by stirring at the same temperature for 3 hours.The The precipitated crystals were collected by filtration, to give after washing with a mixed solvent of THF (100mL) and n- heptane (500mL), and dried under reduced pressure to give the title compound (91.2g). Yield: 72% 1 H-NMR (CDCl 3)δ (ppm): 2.36 (3H, s), 7.49 (1H, dd, J = 2.0,9.5Hz), 7.78 (1H, br), 8.17 (1H, d, J = 2.0Hz). MS (ESI +): 189 [M + 1] +
Step 2: Synthesis of 2-acetylamino-5-chloro-3-fluoro-4-formyl pyridine:
Under a nitrogen atmosphere, and dissolved at room temperature 2-acetylamino-5-chloro-3-fluoropyridine (70.0g, 0.37mol) and 4-formyl-morpholine (128.2g, 1.11mol) to THF (840mL) It was.The solution was cooled to -20 ℃ and was added dropwise a THF solution of 24wt% of lithium hexamethyldisilazide (595g, 0.85mol), and stirred 5.5 hours at the same temperature.The reaction mixture, citric acid monohydrate (257g) and sodium chloride (70g) in an aqueous solution dissolved in water (420mL), and I was added at stirring at 0 ℃.The organic layer was separated and the resulting organic layer was successively washed with 50wt% phosphoric acid aqueous solution of potassium dihydrogen (350mL) and 20wt% sodium chloride solution (350mL) to (1458g).The portion of the organic layer was taken for analysis (292g), and evaporated remainder (1166g) at reduced pressure.The residue in THF (350mL) was added, and the solvent was distilled off under reduced pressure.Again, the residue in THF (350mL) was added to and evaporated under reduced pressure to give a solid (81.4g) containing the title compound.The product was used in the next step without further purification. Some of the organic layer which had been collected (292g) to (29g), and evaporated under reduced pressure.The residue was purified by silica gel column chromatography: subjected to [eluent AcOEt / hexane (1 / 4-9 / 1)], I give the title compound (1.05g, 4.85mmol) as a white powdery solid. Yield: 66% 1 H-NMR (CDCl 3)δ (ppm): 2.40 (3H, s), 7,59 (1H, br), 8.34 (1H, br), 10.42 (1H, s). MS (ESI +): 217 (M + 1)
Under a nitrogen atmosphere to dissolve the solid product of Step 2 (81.4g) in 2,2,2-trifluoroethanol (448mL), piperidine (4.4g, 51.7mmol), acetic acid (3.1g, 51 .7mmol) and 3-oxobutanoic acid ethyl (37.0g, 0.28mol) was added and stirred for 3 hours after raising the temperature to 50 ℃.After cooling the reaction mixture to room temperature, triethylamine (758mL, 5.5mol) and formic acid (172mL, 4.6mol) of 2-propanol (1248mL) solution and 20% Pd (OH) 2 carbon (21.2g, moisture content 46.2%) were added, followed by stirring for 4 hours the temperature was raised to 50 ℃.The reaction mixture was filtered through Celite, and the residue was washed with 2-propanol (679mL).Combined filtrate and washings (2795g), and evaporated under reduced pressure a part of the (399g) (remaining (2396g) I was saved).Ethyl acetate (24.2mL) was added to the residue obtained by evaporation of the solvent, and evaporated under reduced pressure.Again, the residue ethyl acetate (182mL) was added to the washed successively with an organic layer 20wt% brine (61mL), 10wt% of potassium dihydrogen phosphate solution (61mL) and 20wt% sodium chloride solution (61mL), under a reduced pressure The solvent was evaporated.Furthermore, in addition to the residue of 2,2,2-trifluoroethanol (24mL), and the solvent evaporated under reduced pressure to obtain oil containing the title compound (15.0g).The product was used in the next step without further purification. 1 H-NMR (CDCl 3)δ (ppm): 1.24 (3H, t, J = 7.0Hz), 2.27 (3H, s), 2.37 (3H, s), 3.16- 3.26 (2H, m), 3.86 (1H, t, J = 7.5Hz), 4.15-4.22 (2H, m), 6.98 (1H, t, J = 5.0Hz ), 7.68 (1H, br), 8.05 (1H, d, J = 5.0Hz). MS (ESI +): 297 (M + 1)
Step 4: Synthesis of 3- (3-fluoro-2-amino-pyridin-4-ylmethyl) -7-hydroxy-4-methyl-2-oxo -2H-1- benzopyran methanesulphonate:
Under a nitrogen atmosphere, oily product of Step 3 (15.0g) and I were dissolved in 2,2,2-trifluoroethanol (33mL).The solution of resorcinol (5.3g, 47.9mmol) and methane sulfonic acid (11.7mL, 181mmol) was added at 24 ℃, and stirred for 4 hours at 90 ℃.And allowed to stand for 13 hours and cooled to room temperature and ethanol (33mL) and water (11mL), and the mixture was stirred for 4.5 hours at 90 ℃.After adding 2-propanol (105mL) was cooled to 55 ℃, and allowed to stand for 14 hours then cooled to room temperature.The The precipitated crystals were collected by filtration to give 2-propanol was washed twice with (33mL), and dried under reduced pressure to give the title compound (8.2g). (Total from 2-acetylamino-5-chloro-3-fluoropyridine was used in step 2 Yield) Yield: 49% MS (ESI +): 301 [M + 1-MsOH] +
Under a nitrogen atmosphere, 3- (3-fluoro-2-amino-pyridin-4-ylmethyl) -7-hydroxy-4-methyl-2-oxo -2H-1- benzopyran methanesulphonate (7.6g, 19.2mmol) and 2-bromo-pyrimidine (4.0g, 24.9mmol) was dissolved in DMF (122mL), potassium carbonate (5.8g, 42.2mmol) was added, and the mixture was stirred for 3.5 hours at 115 ℃.After cooling the reaction mixture to 28 ℃, water (122mL) was added dropwise over the same temperature for 0.5 hours, and stirred for 2 minutes.In addition, after cooling to 0 ℃, and the mixture was stirred for 1 hour, and the precipitated crystals were collected by filtration.The obtained crystals were washed successively with water (61mL) and acetonitrile (61mL), to give the title compound was dried under reduced pressure and crystals (6.5g). The resultant was taken for analysis a portion of the crystals (0.1g), it was suspended remainder (6.4g) in DMF (70mL).The resulting suspension was stirred 60 ℃ and heated for 5 minutes and stirred for 80 minutes by the addition of acetonitrile (185mL) at the same temperature.Then, it was stirred for 0.5 hours and then cooled to 40 ℃, and the mixture was stirred for 0.5 hours and further cooled to 25 ℃.After a further 1.5 hours with stirring and cooled to 0 ℃, the precipitated crystals were collected by filtration.After washing the resulting crystals in acetonitrile (46mL), was obtained by drying under reduced pressure to the title compound (5.5g).Incidentally, the title compound is a compound described in WO2007 / 091736. Yield: 76%
Under a nitrogen atmosphere, 4-methyl-3- (3-fluoro-2-amino-pyridin-4-ylmethyl) -7- (pyrimidin-2-yloxy) -2-oxo -2H-1- benzopyran (1.7g, 4 the .5mmol) it was suspended in DMF (18mL).To this solution pyridine (0.8mL, 9.9mmol) was cooled to In 10 ℃ added, N- methyl-sulfamoyl chloride (1.05g, 8.1mmol) in acetonitrile (18mL) solution of the internal temperature of 15 ℃ it was dropped so as to maintain below.After stirring for 90 minutes at the same temperature, acetonitrile (3.4mL) was added and further water (50mL), was added dropwise the inner temperature so as to maintain the 20 ℃ below.It was cooled to an external temperature of 0 ℃, and the mixture was stirred for an internal temperature of 5 ℃ 2 hours after arrival.The precipitated crystals were collected by filtration, washed with water (8.5mL), and dried to give the title compound (1.9g, 4.0mmol) was obtained. Yield: 88% MS (ESI +): 472 [M + 1] +
Under a nitrogen atmosphere, 3- {2- (methyl-aminosulfonyl) amino-3-fluoro-pyridin-4-ylmethyl} -4-methyl-7- (pyridin-2-yloxy) -2-oxo -2H-1- benzopyran ( 1.6g, was suspended 3.4mmol) in THF (10mL), water (3mL) was added.The suspension in 2.0M aqueous potassium hydroxide (1.8mL, 3.6mmol) was added dropwise over 10 min at 25 ℃, after raising the temperature to 60 ℃, and the mixture was stirred for 2 hours at the same temperature.After cooling the reaction mixture to 20 ℃, it was added dropwise over a period of THF (8mL) 30 min.After completion of the dropwise addition, the mixture was cooled to an external temperature of -5 ℃, and the mixture was stirred for an internal temperature of 0 ℃ reached after 160 minutes.The precipitated crystals were collected by filtration, then washed with a mixture of THF (14mL) and water (1.6mL) (pre-cooled to 5 ℃), further washed with THF (8mL), and dried to give the title compound (0 .72g, we got 1.4mmol). Yield: 42% MS (ESI +): 472 [M + 2H-K] +
CLIP
RO5126766 (CH5126766) is a first-in-class dual inhibitor of Raf/MEK [1].
The RAS/RAF/MEK/ERK signaling pathway is an important signal transduction system and participates in cell differentiation, movement, division and death. Activated Ras activates RAF kinase, which then phosphorylates and activates MEK (MEK1 and MEK2) [1]. The mutations in BRAF, RAS, and NF1 are associated with many human tumors [2].
RO5126766 (CH5126766) is a first-in-class dual Raf/MEK inhibitor. In cell-free kinase assays, CH5126766 effectively inhibited the phosphorylation of MEK1 protein by RAF and the activation of ERK2 protein by MEK1 with IC50 values of 0.0082-0.056 and 0.16 μM, respectively. In NCI-H460 (KRAS Q61H) human lung large cell carcinoma cell line, RO5126766 induced cell-cycle inhibitor p27Kip1 protein expression and caused G1 arrest. In HCT116 KRAS-mutant colorectal cancer cells, RO5126766 CH5126766 completely inhibited the phosphorylation of MEK and ERK [2].
In Japanese patients with advanced solid tumors, RO5126766 exhibited the maximum tolerable dose (MTD) of 2.25 mg/day once daily [1]. In a HCT116 (G13D KRAS) mouse xenograft model, RO5126766 (1.5 mg/kg) inhibited pERK and ERK signaling and exhibited ED50 value of 0.056 mg/kg [2].
References:
[1]. Honda K, Yamamoto N, Nokihara H, et al. Phase I and pharmacokinetic/pharmacodynamic study of RO5126766, a first-in-class dual Raf/MEK inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol, 2013, 72(3): 577-584.
[2]. Ishii N, Harada N, Joseph EW, et al. Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor, CH5126766, that suppresses feedback reactivation of RAF activity. Cancer Res, 2013, 73(13): 4050-4060.
(From left to right) Principal Investigator Associate Professor Gautam Sethi and NUS PhD candidate Ms Zhang Jingwen from the Department of Pharmacology at the NUS Yong Loo Lin School of Medicine led a research which found that a bioactive compound from the neem plant could significantly suppress development of prostate cancer.
Credit: National University of Singapore
Date:September 29, 2016Source:National University of SingaporeSummary:Oral administration of nimbolide, over 12 weeks shows reduction of prostate tumor size by up to 70 per cent and decrease in tumor metastasis by up to 50 per cent, report investigators.
Oral administration of nimbolide, over 12 weeks shows reduction of prostate tumor size by up to 70 per cent and decrease in tumor metastasis by up to 50 per cent
A team of international researchers led by Associate Professor Gautam Sethi from the Department of Pharmacology at the Yong Loo Lin School of Medicine at the National University of Singapore (NUS) has found that nimbolide, a bioactive terpenoid compound derived from Azadirachta indica or more commonly known as the neem plant, could reduce the size of prostate tumor by up to 70 per cent and suppress its spread or metastasis by half.
Prostate cancer is one of the most commonly diagnosed cancers worldwide. However, currently available therapies for metastatic prostate cancer are only marginally effective. Hence, there is a need for more novel treatment alternatives and options.
“Although the diverse anti-cancer effects of nimbolide have been reported in different cancer types, its potential effects on prostate cancer initiation and progression have not been demonstrated in scientific studies. In this research, we have demonstrated that nimbolide can inhibit tumor cell viability — a cellular process that directly affects the ability of a cell to proliferate, grow, divide, or repair damaged cell components — and induce programmed cell death in prostate cancer cells,” said Assoc Prof Sethi.
Nimbolide: promising effects on prostate cancer
Cell invasion and migration are key steps during tumor metastasis. The NUS-led study revealed that nimbolide can significantly suppress cell invasion and migration of prostate cancer cells, suggesting its ability to reduce tumor metastasis.
The researchers observed that upon the 12 weeks of administering nimbolide, the size of prostate cancer tumor was reduced by as much as 70 per cent and its metastasis decreased by about 50 per cent, without exhibiting any significant adverse effects.
“This is possible because a direct target of nimbolide in prostate cancer is glutathione reductase, an enzyme which is responsible for maintaining the antioxidant system that regulates the STAT3 gene in the body. The activation of the STAT3 gene has been reported to contribute to prostate tumor growth and metastasis,” explained Assoc Prof Sethi. “We have found that nimbolide can substantially inhibit STAT3 activation and thereby abrogating the growth and metastasis of prostate tumor,” he added.
The findings of the study were published in the April 2016 issue of the scientific journal Antioxidants & Redox Signaling. This work was carried out in collaboration with Professor Goh Boon Cher of Cancer Science Institute of Singapore at NUS, Professor Hui Kam Man of National Cancer Centre Singapore and Professor Ahn Kwang Seok of Kyung Hee University.
Neem — The medicinal plant
The neem plant belongs to the mahogany tree family that is originally native to India and the Indian sub-continent. It has been part of traditional Asian medicine for centuries and is typically used in Indian Ayurvedic medicine. Today, neem leaves and bark have been incorporated into many personal care products such as soaps, toothpaste, skincare and even dietary supplements.
Future Research
The team is looking to embark on a genome-wide screening or to perform a large-scale study of proteins to analyse the side-effects and determine other potential molecular targets of nimbolide. They are also keen to investigate the efficacy of combinatory regimen of nimbolide and approved drugs such as docetaxel and enzalutamide for future prostate cancer therapy.
Journal Reference:
Jingwen Zhang, Kwang Seok Ahn, Chulwon Kim, Muthu K. Shanmugam, Kodappully Sivaraman Siveen, Frank Arfuso, Ramar Perumal Samym, Amudha Deivasigamanim, Lina Hsiu Kim Lim, Lingzhi Wang, Boon Cher Goh, Alan Prem Kumar, Kam Man Hui, Gautam Sethi. Nimbolide-Induced Oxidative Stress Abrogates STAT3 Signaling Cascade and Inhibits Tumor Growth in Transgenic Adenocarcinoma of Mouse Prostate Model. Antioxidants & Redox Signaling, 2016; 24 (11): 575 DOI:10.1089/ars.2015.6418
The U.S. Food and Drug Administration today granted accelerated approval to Rubraca (rucaparib) to treat women with a certain type of ovarian cancer. Rubraca is approved for women with advanced ovarian cancer who have been treated with two or more chemotherapies and whose tumors have a specific gene mutation (deleterious BRCA) as identified by an FDA-approved companion diagnostic test.
The U.S. Food and Drug Administration today granted accelerated approval to Rubraca (rucaparib) to treat women with a certain type of ovarian cancer. Rubraca is approved for women with advanced ovarian cancer who have been treated with two or more chemotherapies and whose tumors have a specific gene mutation (deleterious BRCA) as identified by an FDA-approved companion diagnostic test.
“Today’s approval is another example of the trend we are seeing in developing targeted agents to treat cancers caused by specific mutations in a patient’s genes,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research and acting director of the FDA’s Oncology Center of Excellence. “Women with these gene abnormalities who have tried at least two chemotherapy treatments for their ovarian cancer now have an additional treatment option.”
The National Cancer Institute estimates that 22,280 women will be diagnosed with ovarian cancer in 2016 and an estimated 14,240 will die of this disease. Approximately 15 to 20 percent of patients with ovarian cancer have a BRCA gene mutation.
BRCA genes are involved with repairing damaged DNA and normally work to prevent tumor development. However, mutations of these genes may lead to certain cancers, including ovarian cancers. Rubraca is a poly ADP-ribose polymerase (PARP) inhibitor that blocks an enzyme involved in repairing damaged DNA. By blocking this enzyme, DNA inside the cancerous cells with damaged BRCA genes may be less likely to be repaired, leading to cell death and possibly a slow-down or stoppage of tumor growth.
Today, the FDA also approved the FoundationFocus CDxBRCA companion diagnostic for use with Rubraca, which is the first next-generation-sequencing (NGS)-based companion diagnostic approved by the agency. The NGS test detects the presence of deleterious BRCA gene mutations in the tumor tissue of ovarian cancer patients. If one or more of the mutations are detected, the patient may be eligible for treatment with Rubraca.
The safety and efficacy of Rubraca were studied in two, single-arm clinical trials involving 106 participants with BRCA-mutated advanced ovarian cancer who had been treated with two or more chemotherapy regimens. BRCA gene mutations were confirmed in 96 percent of tested trial participants with available tumor tissue using the FoundationFocus CDxBRCA companion diagnostic. The trials measured the percentage of participants who experienced complete or partial shrinkage of their tumors (overall response rate). Fifty-four percent of the participants who received Rubraca in the trials experienced complete or partial shrinkage of their tumors lasting a median of 9.2 months.
Common side effects of Rubraca include nausea, fatigue, vomiting, low levels of red blood cells (anemia), abdominal pain, unusual taste sensation (dysgeusia), constipation, decreased appetite, diarrhea, low levels of blood platelets (thrombocytopenia) and trouble breathing (dyspnea). Rubraca is associated with serious risks, such as bone marrow problems (myelodysplastic syndrome), a type of cancer of the blood called acute myeloid leukemia and fetal harm.
The agency approved Rubraca under its accelerated approval program, which allows approval of a drug to treat a serious or life-threatening disease or condition based on clinical data showing the drug has an effect on a surrogate (substitute) endpoint that is reasonably likely to predict clinical benefit. The sponsor is continuing to study this drug in patients with advanced ovarian cancer who have BRCA gene mutations and in patients with other types of ovarian cancer. The FDA also granted the Rubraca application breakthrough therapy designation and priority review status. Rubraca also received orphan drug designation, which provides incentives such as tax credits, user fee waivers and eligibility for exclusivity to assist and encourage the development of drugs intended to treat rare diseases.
Rubraca is marketed by Clovis Oncology, Inc. based in Boulder, Colorado. The FoundationFocus CDxBRCA companion diagnostic is marketed by Foundation Medicine, Inc. of Cambridge, Massachusetts.
////////////Rubraca, rucaparib, Clovis Oncology, Boulder, Colorado, fda 2016, cancer, ovarian
Approved based on a first-line Phase III trial that met its primary endpoint of progression-free survival (PFS) at interim analysis due to superior efficacy compared to letrozole alone[1]
At this interim analysis, Kisqali plus letrozole reduced risk of disease progression or death by 44% over letrozole alone, and demonstrated tumor burden reduction with a 53% overall response rate[1]
Kisqali plus letrozole showed treatment benefit across all patient subgroups regardless of disease burden or tumor location[1]
At a subsequent analysis with additional follow-up and progression events, a median PFS of 25.3 months for Kisqali plus letrozole and 16.0 months for letrozole alone was observed[2]
Basel, March 13, 2017– The US Food and Drug Administration (FDA) has approved Kisqali®(ribociclib, formerly known as LEE011) in combination with an aromatase inhibitor as initial endocrine-based therapy for treatment of postmenopausal women with hormone receptor positive, human epidermal growth factor receptor-2 negative (HR+/HER2-) advanced or metastatic breast cancer.
Kisqali is a CDK4/6 inhibitor approved based on a first-line Phase III trial that met its primary endpoint early, demonstrating statistically significant improvement in progression-free survival (PFS) compared to letrozole alone at the first pre-planned interim analysis[1]. Kisqali was reviewed and approved under the FDA Breakthrough Therapy designation and Priority Review programs.
“Kisqali is emblematic of the innovation that Novartis continues to bring forward for people with HR+/HER2- metastatic breast cancer,” said Bruno Strigini, CEO, Novartis Oncology. “We at Novartis are proud of the comprehensive clinical program for Kisqali that has led to today’s approval and the new hope this medicine represents for patients and their families.”
The FDA approval is based on the superior efficacy and demonstrated safety of Kisqali plus letrozole versus letrozole alone in the pivotal Phase III MONALEESA-2 trial. The trial, which enrolled 668 postmenopausal women with HR+/HER2- advanced or metastatic breast cancer who received no prior systemic therapy for their advanced breast cancer, showed that Kisqali plus an aromatase inhibitor, letrozole, reduced the risk of progression or death by 44 percent over letrozole alone (median PFS not reached (95% CI: 19.3 months-not reached) vs. 14.7 months (95% CI: 13.0-16.5 months); HR=0.556 (95% CI: 0.429-0.720); p<0.0001)[1].
More than half of patients taking Kisqali plus letrozole remained alive and progression free at the time of interim analysis, therefore median PFS could not be determined[1]. At a subsequent analysis with additional 11-month follow-up and progression events, a median PFS of 25.3 months for Kisqali plus letrozole and 16.0 months for letrozole alone was observed[2]. Overall survival data is not yet mature and will be available at a later date.
“In the MONALEESA-2 trial, ribociclib plus letrozole reduced the risk of disease progression or death by 44 percent over letrozole alone, and more than half of patients (53%) with measurable disease taking ribociclib plus letrozole experienced a tumor burden reduction of at least 30 percent. This is a significant result for women with this serious form of breast cancer,” said Gabriel N. Hortobagyi, MD, Professor of Medicine, Department of Breast Medical Oncology, The University of Texas MD Anderson Cancer Center and MONALEESA-2 Principal Investigator. “These results affirm that combination therapy with a CDK4/6 inhibitor like ribociclib and an aromatase inhibitor should be a new standard of care for initial treatment of HR+ advanced breast cancer.”
Kisqali is taken with or without food as a once-daily oral dose of 600 mg (three 200 mg tablets) for three weeks, followed by one week off treatment. Kisqali is taken in combination with four weeks of any aromatase inhibitor[1].
Breast cancer is the second most common cancer in American women[3]. The American Cancer Society estimates more than 250,000 women will be diagnosed with invasive breast cancer in 2017[3]. Up to one-third of patients with early-stage breast cancer will subsequently develop metastatic disease[4].
Novartis is committed to providing patients with access to medicines, as well as resources and support to address a range of needs. The Kisqali patient support program is available to help guide eligible patients through the various aspects of getting started on treatment, from providing educational information to helping them understand their insurance coverage and identify potential financial assistance options. For more information, patients and healthcare professionals can call 1-800-282-7630.
About Kisqali® (ribociclib)
Kisqali (ribociclib) is a selective cyclin-dependent kinase inhibitor, a class of drugs that help slow the progression of cancer by inhibiting two proteins called cyclin-dependent kinase 4 and 6 (CDK4/6). These proteins, when over-activated, can enable cancer cells to grow and divide too quickly. Targeting CDK4/6 with enhanced precision may play a role in ensuring that cancer cells do not continue to replicate uncontrollably.
Kisqali was developed by the Novartis Institutes for BioMedical Research (NIBR) under a research collaboration with Astex Pharmaceuticals.
About the MONALEESA Clinical Trial Program
Novartis is continuing to assess Kisqali through the robust MONALEESA clinical trial program, which includes two additional Phase III trials, MONALEESA-3 and MONALEESA-7, that are evaluating Kisqali in multiple endocrine therapy combinations across a broad range of patients, including premenopausal women. MONALEESA-3 is evaluating Kisqali in combination with fulvestrant compared to fulvestrant alone in postmenopausal women with HR+/HER2- advanced breast cancer who have received no or a maximum of one prior endocrine therapy. MONALEESA-7 is investigating Kisqali in combination with endocrine therapy and goserelin compared to endocrine therapy and goserelin alone in premenopausal women with HR+/HER2- advanced breast cancer who have not previously received endocrine therapy.
About Novartis in Advanced Breast Cancer
For more than 25 years, Novartis has been at the forefront of driving scientific advancements for breast cancer patients and improving clinical practice in collaboration with the global community. With one of the most diverse breast cancer pipelines and the largest number of breast cancer compounds in development, Novartis leads the industry in discovery of new therapies and combinations, especially in HR+ advanced breast cancer, the most common form of the disease.
Kisqali® (ribociclib) Important Safety Information
Kisqali® (ribociclib) can cause a heart problem known as QT prolongation. This condition can cause an abnormal heartbeat and may lead to death. Patients should tell their healthcare provider right away if they have a change in their heartbeat (a fast or irregular heartbeat), or if they feel dizzy or faint. Kisqali can cause serious liver problems. Patients should tell their healthcare provider right away if they get any of the following signs and symptoms of liver problems: yellowing of the skin or the whites of the eyes (jaundice), dark or brown (tea-colored) urine, feeling very tired, loss of appetite, pain on the upper right side of the stomach area (abdomen), and bleeding or bruising more easily than normal. Low white blood cell counts are very common when taking Kisqali and may result in infections that may be severe. Patients should tell their healthcare provider right away if they have signs and symptoms of low white blood cell counts or infections such as fever and chills. Before taking Kisqali, patients should tell their healthcare provider if they are pregnant, or plan to become pregnant as Kisqali can harm an unborn baby. Females who are able to become pregnant and who take Kisqali should use effective birth control during treatment and for at least 3 weeks after the last dose of Kisqali. Do not breastfeed during treatment with Kisqali and for at least 3 weeks after the last dose of Kisqali. Patients should tell their healthcare provider about all of the medicines they take, including prescription and over-the-counter medicines, vitamins, and herbal supplements since they may interact with Kisqali. Patients should avoid pomegranate or pomegranate juice, and grapefruit or grapefruit juice while taking Kisqali. The most common side effects (incidence >=20%) of Kisqali when used with letrozole include white blood cell count decreases, nausea, tiredness, diarrhea, hair thinning or hair loss, vomiting, constipation, headache, and back pain. The most common grade 3/4 side effects in the Kisqali + letrozole arm (incidence >2%) were low neutrophils, low leukocytes, abnormal liver function tests, low lymphocytes, and vomiting. Abnormalities were observed in hematology and clinical chemistry laboratory tests.
About Novartis
Novartis provides innovative healthcare solutions that address the evolving needs of patients and societies. Headquartered in Basel, Switzerland, Novartis offers a diversified portfolio to best meet these needs: innovative medicines, cost-saving generic and biosimilar pharmaceuticals and eye care. Novartis has leading positions globally in each of these areas. In 2016, the Group achieved net sales of USD 48.5 billion, while R&D throughout the Group amounted to approximately USD 9.0 billion. Novartis Group companies employ approximately 118,000 full-time-equivalent associates. Novartis products are sold in approximately 155 countries around the world. For more information, please visit http://www.novartis.com.
References
[1] Kisqali (ribociclib) Prescribing information. East Hanover, New Jersey, USA: Novartis Pharmaceuticals Corporation; March 2016.
[2] Novartis Data on File
[3] American Cancer Society. How Common Is Breast Cancer? Available at https://www.cancer.org/cancer/breast-cancer/about/how-common-is-breast-cancer.html(link is external). Accessed January 23, 2017.
[4] O’Shaughnessy J. Extending survival with chemotherapy in metastatic breast cancer. The Oncologist. 2005;10(Suppl 3):20-29.
The Food and Drug Administration (FDA) has approved several quinazoline derivatives for clinical use as anticancer drugs. These include gefitinib, erlotinib, lapatinib, afatinib, and vandetanib (Fig.1) [43]. Gefitinib (Iressa®) was approved by the FDA in 2003 for the treatment of locally advanced or metastatic non-small-cell lung cancer (NSCLC) in patients after failure of both platinum-based and/or docetaxel chemotherapies. In 2004, erlotinib (Tarceva®) was approved by the FDA for treating NSCLC. Furthermore, in 2005, the FDA approved erlotinib in combination with gemcitabine for treatment of locally advanced, unrespectable, or metastatic pancreatic cancer. Erlotinib acts as a reversible tyrosine kinase inhibitor. Lapatinib (Tykreb®) was approved by the FDA in 2012 for breast cancer treatment. It inhibits the activity of both human epidermal growth factor receptor-2 (HER2/neu) and epidermal growth factor receptor (EGFR) pathways. Vandetanib (Caprelsa®) was approved by the FDA in 2011 for the treatment of metastatic medullary thyroid cancer. It acts as a kinase inhibitor of a number of cell receptors, mainly the vascular endothelial growth factor receptor (VEGFR), EGFR, and rearranged during transfection (RET)-tyrosine kinase (TK). Afatinib (Gilotrif®) was approved by the FDA in 2013 for NSCLC treatment. It acts as an irreversible covalent inhibitor of the receptor tyrosine kinases (RTK) for EGFR and erbB-2 (HER2).
aDepartment of Mathematics and Natural Sciences, School of Arts and Sciences, American University of Ras Al Khaimah, Ras Al Khaimah, United Arab Emirates E-mail:shagufta.waseem@aurak.ac.ae, iahmad@aurak.ac.ae
Abstract
Cancer is one of the major causes of worldwide human mortality. A wide range of cytotoxic drugs are available on the market, and several compounds are in different phases of clinical trials. Many studies suggest that these cytotoxic molecules are also associated with different types of adverse side effects; therefore researchers around the globe are involved in the development of more efficient and safer anticancer drugs. In recent years, quinazoline and its derivatives have been considered as a novel class of cancer chemotherapeutic agents that show promising activity against different tumors. The aim of this article is to comprehensively review and highlight the recent developments concerning the anticancer activity of quinazoline derivatives as well as offer perspectives on the development of novel quinazoline derivatives as anticancer agents in the near future.
Dr. Shagufta joined the American University of Ras Al Khaimah as an Assistant Professor of Chemistry in the School of Arts and Sciences in August 2014. Prior to joining AURAK, Dr. Shagufta worked as an Adjunct Assistant Professor of Chemistry at the University of Modern Sciences, Dubai and American University of Ras Al Khaimah, UAE.
Dr. Shagufta also worked as a Postdoctoral Researcher Associate at the Department of Chemistry and Biochemistry, Oklahoma University, USA. She developed the noble drug delivery system for breast cancer drugs using carbon nanotubes and acquired the significant experience in nanotechnology and synthetic organic chemistry. She was appointed as a Postdoctoral Research Fellow and Visiting Scientist at Leiden/Amsterdam Centre for Drug Research (LACDR), Leiden, The Netherlands. Her research interest was In silico prediction and clinical evaluation of the cardiotoxicity of drug candidates. She was focused to identify chemical substructures as ‘chemical alerts’ that interact with this hERG channel. Dr. Shagufta received a Ph.D. under the prestigious CSIR-JRF and SRF research fellowship in Chemistry from Central Drug Research Institute (CDRI)/Lucknow University, India in 2008, her PhD research work was in the field of estrogens and antiestrogens, design and synthesis of steroidal and non-steroidal tissue selective estrogen receptor modulators (SERMs) for breast cancer, 3D-QSAR CoMFA and CoMSIA studies and analysis of pharmaceutical important molecules.
Dr. Shagufta has published 20 articles in peer-reviewed International journals of Royal Society of Chemistry, Elsevier, Wiley and Springer. Dr. Shagufta teaches courses such as General chemistry, Organic Chemistry, Chemistry in Everyday Life, and Spectroscopy along with laboratory courses.
Research and Publication
Research Interest-Dr. Shagufta
Organic Chemistry, Medicinal Chemistry focused on Breast Cancer and Osteoporosis, Heterogeneous catalysis and Nanotechnology.
Publications- Dr. Shagufta
Irshad Ahmad and Shagufta. 2015. Recent developments in steroidal and nonsteroidal aromatase inhibitors for the chemoprevention of estrogen-dependent breast cancer. European Journal of Medicinal Chemistry, 102, 375-386.
Irshad Ahmad and Shagufta. 2015. Sulfones: An important class of organic compounds with diverse biological activities. International Journal of Pharmacy and Pharmaceutical Sciences, 7 (3), 19-27.
Donna J. Nelson, Shagufta, Ravi Kumar. 2012. Characterization of a tamoxifen-tethered single-walled carbon nanotube conjugate by using NMR spectroscopy. Anal. Bioanal. Chem.[Springer] 404:771–776. [ISSN: 1618-2642]
Donna J. Nelson, Ravi Kumar, Shagufta. 2012. Regiochemical reversals in nitrosobenzene reactions with carbonyl compounds – α-aminooxy ketone versus α-hydroxyamino ketone products. Eur. J. Org. Chem.(Wiley-VCH) 6013-6020. [ISSN: 1099-0690]
Munikumar R. Reddy, Elisabeth Klaasse, Shagufta, Adriaan P. IJzerman, Andreas Bender. 2010. Validation of an in silico hERG model and its applications to the virtual screening of commercial compound databases. Chem. Med. Chem. (Wiley-VCH)5: 716-729. [ISSN: 1860-7187]
Shagufta, Dong Guo, Elisabeth Klaasse, Henk de Vries, Johannes Brussee, Lukas Nalos, Martin B Rook, Marc A Vos, Marcel AG van der Heyden and Adriaan P. IJzerman. 2009. Exploring the chemical substructures essential for hERG K+ channel blockade by synthesis and biological evaluation of dofetilide analogues. Chem. Med. Chem.(Wiley-VCH) 4:1722-1732. [ISSN: 1860-7187]
Shagufta, Ritesh Singh and Gautam Panda. 2009, Synthetic studies towards steroid-amino acid hybrids. Indian Journal of Chemistry.(Indian Science) 48B: 989-995. [ISSN: 0975-0983]
Maloy K. Parai, Shagufta, Ajay K. Srivastava, Matthias Kassack, Gautam Panda. 2008. An unexpected reaction of phosphorous tribromide on chromanone, thiochromanone, 3,4-dihydro-2H-benzo[b]thiepin-5-one, 3,4-dihydro-2H-benzo[b]oxepin-5-one and tetralone derived allylic alcohols: a case study. Tetrahedron (Elsevier)64: 9962-9976. [ISSN: 0040-4020]
Gautam Panda, Maloy Kumar Parai, Sajal Kumar Das, Shagufta, Manish Sinha, Vinita Chaturvedi, Anil K. Srivastava, Anil N. Gaikwad, Sudhir Sinha. 2007. Effect of substituents on diarymethanes for antitubercular activity. European Journal of Medicinal Chemistry (Elsevier) 42: 410-419. [ISSN: 0223-5234]
Shagufta and Gautam Panda. 2007. A new example of a steroid-amino acid hybrid: Construction of constrained nine membered D-ring steroids. Organic and Biomolecular Chemistry (Royal Society of Chemistry) 5 : 360- 366. [ISSN 1477-0539]
Shagufta, Ashutosh Kumar, Gautam Panda and Mohammad Imran Siddiqi. 2007. CoMFA and CoMSIA 3D-QSAR analysis of diaryloxy methano phenanthrene derivatives as anti- tubercular agents. Journal of Molecular Modeling (Springer) 13: 99-107. [ISSN:0948-5023]
Shagufta, Ajay Kumar Srivastava, Ramesh Sharma, Rajeev Mishra, Anil K. Balapure, Puvvada S. R. Murthy and Gautam Panda. 2006. Substituted phenanthrenes with basic amino side chains: A new series of anti-breast cancer agents. Bioorganic and Medicinal Chemistry (Elsevier) 14: 1497-1505. [ISSN: 0968-0896]
Shagufta, Ajay Kumar Srivastava and Gautam Panda. 2006. Isomerization of allylic alcohols into saturated carbonyls using phosphorus tribromide. Tetrahedron Letters (Elsevier) 47: 1065-1070. [ISSN: 0040-4039]
Gautam Panda, Jitendra K. Mishra, Shagufta, T. C. Dinadayalane and G. Narahari Sastry & Devendra S Negi. 2006. Hard-soft acid-base (HSAB) principle and difference in d-orbital configurations of metals explain the regioselectivity of nucleophilic attack to a carbinol in Friedel-Crafts reaction catalyzed by Lewis and protonic acids. Indian Journal of Chemistry (Indian Science)45B: 276-287. [ISSN: 0975-0983]
Shagufta, Maloy Kumar Parai and Gautam Panda. 2005. A new strategy for the synthesis of aryl- and heteroaryl-substituted exocyclic olefins from allyl alcohols using PBr3. Terahedron Letters (Elsevier) 46: 8849-8852. [ISSN: 0040-4039]
Shagufta, Resmi Raghunandan, Prakash R. Maulik and Gautam Panda. 2005. Convenient phosphorus tribromide induced syntheses of substituted 1-arylmethylnaphthalenes from 1-tetralone derivatives. Tetrahedron Letters (Elsevier) 46: 5337-5341. [ISSN: 0040-4039]
Gautam Panda, Shagufta, Anil K. Srivastava and Sudhir Sinha. 2005. Synthesis and antitubercular activity of 2-hydroxy-aminoalkyl derivatives of diaryloxy methano phenanthrenes. Bioorganic and Medicinal Chemistry Letters (Elsevier) 15: 5222-5225. [ISSN: 0960-894X]
Sajal Kumar Das, Shagufta, and Gautam Panda. 2005. An easy access to unsymmetric trisubstituted methane derivatives (TRSMs). Tetrahedron Letters (Elsevier) 46: 3097-3102. [ISSN: 0040-4039]
Shagufta, Jitendra Kumar Mishra, Vinita Chaturvedi, Anil K. Srivastava, Ranjana Srivastava and Brahm S. Srivastava. 2004. Diaryloxy methano phenanthrenes: a new class of antituberculosis agents. Bioorganic and Medicinal Chemistry (Elsevier) 12: 5269-5276. [ISSN: 0968-0896]
Dr. Irshad Ahmad joined the American University of Ras Al Khaimah in spring 2011 as an Assistant Professor of Chemistry. He received the master’s degree in chemistry from Jiwaji University in 1999. Subsequently acquired significant pharmaceutical industrial experience and developed cardio-selective beta-blocker drug molecule. He joined Central Salt and Marine Chemical Research Institute and Bhavnagar University under the sponsored project of DST and CSIR as a senior research fellow and received his PhD degree in chemistry in 2006. Subsequently, he accepted an invited scientist position in Korea Research Institute of Chemical Technology, South Korea and contributed his expertise in the field of Nanotechnology. Dr. Irshad is a recipient of prestigious European fellowships (NWO-Rubicon & FCT) and he joined Van’t Hoff Institute for Molecular Sciences, University of Amsterdam, The Netherlands as a NWO Rubicon fellow (Netherlands Organization for Scientific Research, the Dutch Science Foundation), he acquired expertise in the field of supramolecular chemistry.
Afterward, he moved to the Leibniz Institute for Surface Modification, Leipzig, Germany under the Deutsche Forschungsgemeinschaft Grant. Dr. Irshad developed “Novel ultra-fast metathesis catalyst” for the production of high quality alternating copolymers. Subsequently Dr. Irshad, joined Department of Chemistry and Biochemistry, Stephenson Life Science Research Center, University of Oklahoma, USA as a postdoctoral research associate. He developed strategies for the novel environmentally friendly reactions for the production of value added chemicals from biomass.
Dr. Irshad specialized in the area of chemistry, bridging the traditional disciplines of inorganic, organic and bio-organic chemistry. He contributed US and European patent for green and clean technology development. He has published peer-reviewed international research articles in the American Chemical Society (ACS), Royal Society of Chemistry (RSC) Cambridge, Elsevier Science, Wiley, and Springer journals. He has presented his research at several scientific conferences worldwide and received awards.
Research and Publication
Research Interest:
Asymmetric catalysis, Biotechnology, Metathesis, Material science, Nanotechnology, Pharmaceutical, Renewable energy and Supramolecular chemistry
Book:
Asymmetric Homogeneous and Heterogeneous Catalysts: An Approach to the Synthesis of Chiral Drug Intermediates by Scholars Press, Germany. 2013, ISBN: 978-3-639-51138-3
Membership:
American Chemical Society (ACS), USA
The Royal Society of Chemistry, Cambridge, UK
Patents:
United States Patent 7,235,676, H. Khan, S. H. R. Abdi, R. I. Kureshy, S. Singh, I. Ahmad, R. V. Jasra, P. K. Ghosh, ‘Catalytic process for the preparation of epoxides from alkenes.
Patent Cooperation Treaty (PCT) WO/2005/095370, N. H. Khan, S. H. R. Abdi, R. I. Kureshy, S. Singh, I. Ahmad, R. V. Jasra, P. K. Ghosh. An improved catalytic process for the preparation of epoxides from alkenes.
European Patent EP 1732910 A1, N. H. Khan, S. H. R. Abdi, R. I. Kureshy, S. SinghA, I. Ahmad, R. V. Jasra, P. K. Ghosh, An improved catalytic process for the preparation of epoxides from alkenes.
Publications:
Pramoda, U. Gupta, I. Ahmad, R. Kumar, C.N.R. Rao, Assemblies of Covalently Cross-linked Nanosheets of MoS2 and of MoS2-RGO: Synthesis and Novel Properties, Journal of Materials Chemistry A, 4, 2016, 8989.
Shagufta, I. Ahmad, Recent insight into the biological activities of synthetic xanthone derivatives, European Journal of Medicinal Chemistry, 116, 2016, 267.
Ahmad, Shagufta, Recent Development in Steroidal and Non-steroidal Aromatase Inhibitors for the Chemoprevention of Estrogen dependent Breast Cancer, European Journal of Medicinal Chemistry, 102, 2015, 375.
Ahmad, Shagufta, Sulfones: An important class of organic compounds with diverse biological activities, International Journal of Pharmacy and Pharmaceutical Sciences, 7, 3, 2015, 19.
Kumar, K. Gopalakrishnan, I. Ahmad, and C. N. R. Rao, BN-Graphene Composites Generated by Covalent Cross-Linking with Organic Linkers, Advanced Functional Materials, 25, 37, 2015, 5910.
Kumar, D. Raut, I. Ahmad, U. Ramamurty, T. K. Maji and C. N. R. Rao. Functionality preservation with enhanced mechanical integrity in the nanocomposites of the metal–organic framework, ZIF-8, with BN nanosheets, Materials Horizons, 1, 2014, 513.
R. Buchmeiser, I. Ahmad, V. Gurram and P. S. Kumar, Pseudo-Halide and Nitrate Derivatives of Grubbs and Grubbs_Hoveyda Initiators: Some Structural Features Related to the Alternating Ring-Opening Metathesis Copolymerization of Norborn-2-ene with Cyclic Olefins, Macromolecule, 44 (11), 2011, 4098.
Ahmad, G. Chapman and K. M. Nicholas, Sulfite-Driven, Oxorhenium-Catalyzed Deoxydehydration of Glycols, Organometallics, 30 (10), 2011, 2810.
Vkuturi, G. Chapman, I. Ahmad, K. M. Nicholas, Rhenium-Catalyzed Deoxydehydration of Glycols by Sulfite, Inorganic Chemistry, 49, 2010, 4744.
I. Kureshy, I. Ahmad, K. Pathak, N. H. Khan, S. H. R. Abdi, H. C. Bajaj, Solvent- free microwave synthesis of aryloxypropanolamines by ring opening of aryloxy epoxides, Research Letters in Organic Chemistry, 2009, Article ID 109717, doi:10.1155/2009/109717.
I. Kureshy, I. Ahmad, K. Pathak, N. H. Khan, S. H. R. Abdi, R. V. Jasra, Sulfonic acid functionalized mesoporous SBA-15 as an efficient and recyclable catalyst for the synthesis of chromenes from chromanols, Catalysis Communications 10, 2009, 572.
Pathak, I. Ahmad, S. H. R. Abdi, R. I. Kureshy, N. H. Khan, R. V. Jasra, The synthesis of silica-supported chiral BINOL: Application in Ti-catalyzed asymmetric addition of diethylzinc to aldehydes, Journal of Molecular Catalysis A-Chemical 280, 2008, 106.
Kluwer, I. Ahmad, J. N. H. Reek, Improved synthesis of monodentate and bidentate 2- and 3-pyridylphosphines, Tetrahedron Letter 48, 2007, 2999.
Pathak, I. Ahmad, S. H. R. Abdi, R. I. Kureshy, N. H. Khan, R. V. Jasra, Oxidative Kinetic Resolution of racemic Secondary Alcohols catalyzed by recyclable Dimeric Mn(III) salen catalysts, Journal of Molecular Catalysis A-Chemical 274, 2007, 120.
I. Kureshy, I. Ahmad, N. H. Khan, S. H. R. Abdi, K. Pathak, R. V. Jasra, Easily Recyclable Chiral Polymeric Mn (salen) Complex for Oxidative Kinetic resolution of Racemic Secondary Alcohols, Chirality, 19, 2007, 352.
Pathak, A. P. Bhatt, S. H. R. Abdi, R. I. Kureshy, N. H. Khan, I. Ahmad, R. V. Jasra, Enantioselective phenylacetylene addition to aromatic aldehydes and ketones catalyzed by recyclable polymeric Zn(II) salen complex, Chirality, 19, 2007, 1.
I. Kureshy, I. Ahmad, N. H. Khan, S. H. R. Abdi, K. Pathak, R. V. Jasra, Chiral Mn (III) salen complexes covalently bonded on modified MCM-41 and SBA-15 as efficient catalysts for enantioselective epoxidation of non- functionalized alkenes, Journal of Catalysis A-Chemical, 238, 2006, 134.
Pathak, A. P. Bhatt, S. H. R. Abdi, R. I. Kureshy, N. H. Khan, I. Ahmad, R. V. Jasra Enantioselective addition of diethylzinc to aldehydes using immobilized chiral BINOL-Ti complex on ordered mesoporous silicas, Tetrahedron: Asymmetry,17, 2006, 1506.
I. Kureshy, I. Ahmad, N. H. Khan, S. H. R. Abdi, K. Pathak, R. V. Jasra, Encapsulation of chiral MnIII (salen) complex in ordered mesoporous silicas: An approach Towards heterogenizing asymmetric Epoxidation catalysts for non-Functionalized alkenes, Tetrahedron: Asymmetry 16, 2005, 3562.
I. Kureshy, I. Ahmad, N. H. Khan, S. H. R. Abdi, S. Singh, P. H. Pandia, R. V. Jasra, New immobilized chiral Mn(III) salen complexes on pyridine N-Oxide Modified MCM-41as effective catalysts for epoxidation of nonfunctionalized Alkenes, Journal of Catalysis A- Chemical 235 , 2005, 28.
Pathak, A. P. Bhatt, S. H. R. Abdi, R. I. Kureshy, N. H. Khan, I. Ahmad, R. V. Jasra Enantioselective addition of diethylzinc to aldehydes using immobilized chiral BINOL-Ti complex on ordered mesoporous silicas, Tetrahedron: Asymmetry,17, 2006, 1506.
I. Kureshy, S. Singh, N. H. Khan, S. H. R. Abdi, I. Ahmad, A. Bhatt, R. V. Jasra, Improved catalytic activity of homochiral dimeric cobalt salen hydrolytic kinetic resolution of terminal racemic epoxides, Chirality, 17, 2005, 1.
I. Kureshy, S. Singh, N. H. Khan, S. H. R. Abdi , I. Ahmad, .Bhatt, R. V. Jasra, Environment friendly protocol for enantioselective epoxidation of non-functionalized Alkenes catalyzed by recyclable homochiral dimeric Mn(III)salen complexes with hydrogen peroxide and UHP adduct as Oxidants, Catalysis Letters, 107, 2005, 127.
I. Kureshy, N. H. Khan, S. H. R. Abdi, I. Ahmad, S. Singh, and R. V. Jasra, Dicationic chiral Mn (III) Salen complex exchange in the interlayers of Montmorillonite clay: a heterogeneous enantioselective catalyst for epoxidation of non-functionalised alkenes, Journal of Catalysis, 221, 2004, 234.
I. Kureshy, N. H. Khan, S. H. R. Abdi, S. Singh, I. Ahmad, R. V. Jasra, Catalytic asymmetric epoxidation of non-functionalised alkenes using polymeric Mn(III)Salen as catalysts and NaOCl as oxidant, Journal of Molecular Catalysis A-Chemical, 218, 2004, 141.
I. Kureshy, N.H. Khan, S.H. R. Abdi, A. P. Vyas, I. Ahmad, S. Singh, R. V. Jasra, Enantioselective Epoxidation of Non-Functionalised Alkenes catalysed by recyclable new Homo Chiral Dimeric Mn(III) Salen complexes, Journal of Catalysis, 224, 2004, 229.
I. Kureshy, N. H. Khan, S. H. R. Abdi, I. Ahmad, S. Singh, and R. V. Jasra, Immobilization of dicationic Mn(III) salen in the interlayers of montmorrillonite Clay for enantioselective epoxidation of non-functionalised alkenes, Catalysis Letters, 91, 2003, 207.
Selected International Events:
Applied Nanotechnology and Nanoscience International Conference (ANNIC), November 9-11, 2016, Barcelona, SPAIN.
2nd International Conference on Smart Material Research (ICSMR), September 22-24, 2016, Istanbul, TURKEY.
Emirates Foundation’s Think Science Competition, April 17-19, 2016, World Trade Center, Dubai, UAE.
SSL Visiting Fellow 2013-15 at the International Centre for Materials Science, JNCASR, SSL, Bangalore, INDIA.
Global Conference on Materials Sciences (GC-MAS-2014), November 13-15, 2014, Antalya, TURKEY.
5th Annual International Workshop on Advanced Material (IWAM 2013), organized by Ras Al Khaimah Center for Advance Materials (RAK CAM), Feb. 24-26, 2013 at Al Hamra Fort Hotel, Ras Al Khaimah, UAE.
Internal Quality Assurance in Higher Education Institutions workshop organized by the Commission for Academic Accreditation (CAA)- 2nd May 2011, Alghurair University campus, Dubai, UAE.
45th American Chemical Society (ACS) Midwest Regional meeting, Oct. 27-30, 2010, Wichita, Kansas, USA.
55th Annual American Chemical Society (ACS) PentaSectional Meeting- Biofuel, April 10, 2010, organized by American Chemical Society (ACS), Norman, Oklahoma, USA.
18th International Symposium on Olefin Metathesis and Related Chemistry (ISOM XVIII), Organized by the Leibniz-Institute for Surface modification (IOM), August 2-7, 2009, Leipzig, GERMANY.
16th International Symposium on Homogeneous Catalysis (ISHC-XVI), July 6-11, 2008, Organized by the Institute of Chemistry of Organometallic Compounds (ICCOM) of the Italian Research Council (CNR) held in Florence, ITALY.
European IDECAT Summer School on Computational Methods for Catalysis and Materials Science, 15-22 September 2007, Porquerolles, FRANCE.
8th Netherland’s Catalysis and Chemistry Conference (NCCC), March 5-7, 2007, Noordwijkerhout, The NETHERLANDS.
7th International Symposium on Catalysis Applied to Fine Chemicals organized by German Catalysis Society and Dechema. Oct 23-27, 2005, Bingen -Mainz, GERMANY.
1st Indo- German Conference on Catalysis-A Cross Disciplinary Vision, February 6-8, 2003, Indian Institute of Chemical Technology (IICT), Hyderabad, INDIA.
LY2606368 is a small-molecule Chk-1 inhibitors invented by Array and being developed by Eli Lilly and Company. Lilly is responsible for all clinical development and commercialization activities. Chk-1 is a protein kinase that regulates the tumor cell’s response to DNA damage often caused by treatment with chemotherapy. In response to DNA damage, Chk-1 blocks cell cycle progression in order to allow for repair of damaged DNA, thereby limiting the efficacy of chemotherapeutic agents. Inhibiting Chk-1 in combination with chemotherapy can enhance tumor cell death by preventing these cells from recovering from DNA damage.
Originator Array BioPharma; Eli Lilly
Developer Eli Lilly; National Cancer Institute (USA)
Class Antineoplastics; Nitriles; Pyrazines; Pyrazoles; Small molecules
Phase II Breast cancer; Ovarian cancer; Small cell lung cancer; Solid tumours
Phase I Acute myeloid leukaemia; Colorectal cancer; Head and neck cancer; Myelodysplastic syndromes; Non-small cell lung cancer
Most Recent Events
10 Apr 2017 Eli Lilly completes a phase I trial for Solid tumours (Late-stage disease, Second-line therapy or greater) in Japan (NCT02514603)
10 Mar 2017 Phase-I clinical trials in Solid tumours (Combination therapy, Metastatic disease, Inoperable/Unresectable) in USA (IV) (NCT03057145)
22 Feb 2017 Khanh Do and AstraZeneca plan a phase H trial for Solid tumour (Combination therapy, Metastatic disease, Inoperable/Unresectable) in USA (NCT03057145)
Prexasertib (LY2606368) is a small molecule checkpoint kinase inhibitor, mainly active against CHEK1, with minor activity against CHEK2. This causes induction of DNA double-strand breaks resulting in apoptosis. It is in development by Eli Lilly. [1]
A phase II clinical trial for the treatment of small cell lung cancer is expected to be complete in December 2017.[2]
an aminopyrazole compound, or a pharmaceutically acceptable salt thereof or a solvate of the salt, that inhibits Chkl and is useful for treating cancers characterized by defects in deoxyribonucleic acid (DNA) replication, chromosome segregation, or cell division.
Chkl is a protein kinase that lies downstream from Atm and/or Atr in the DNA damage checkpoint signal transduction pathway. In mammalian cells, Chkl is phosphorylated in response to agents that cause DNA damage including ionizing radiation (IR), ultraviolet (UV) light, and hydroxyurea. This phosphorylation which activates Chkl in mammalian cells is dependent on Atr. Chkl plays a role in the Atr dependent DNA damage checkpoint leading to arrest in S phase and at G2M. Chkl phosphorylates and inactivates Cdc25A, the dual-specificity phosphatase that normally dephosphorylates cyclin E/Cdk2, halting progression through S-phase. Chkl also phosphorylates and inactivates Cdc25C, the dual specificity phosphatase that dephosphorylates cyclin B/Cdc2 (also known as Cdkl) arresting cell cycle progression at the boundary of G2 and mitosis (Fernery et al, Science, 277: 1495-1, 1997). In both cases, regulation of Cdk activity induces a cell cycle arrest to prevent cells from entering mitosis in the presence of DNA damage or unreplicated DNA. Various inhibitors of Chkl have been reported. See for example, WO 05/066163,
WO 04/063198, WO 03/093297 and WO 02/070494. In addition, a series of aminopyrazole Chkl inhibitors is disclosed in WO 05/009435.
However, there is still a need for Chkl inhibitors that are potent inhibitors of the cell cycle checkpoints that can act effectively as potentiators of DNA damaging agents. The present invention provides a novel aminopyrazole compound, or a pharmaceutically acceptable salt thereof or solvate of the salt, that is a potent inhibitor of Chkl . The compound, or a pharmaceutically acceptable salt thereof or a solvate of the salt, potently abrogates a Chkl mediated cell cycle arrest induced by treatment with DNA damaging agents in tissue culture and in vivo. Furthermore, the compound, or a pharmaceutically acceptable salt thereof or a solvate of the salt, of the present invention also provides inhibition of Chk2, which may be beneficial for the treatment of cancer. Additionally, the lack of inhibition of certain other protein kinases, such as CDKl, may provide a -2- therapeutic benefit by minimizing undesired effects. Furthermore, the compound, or a pharmaceutically acceptable salt thereof or a solvate of the salt, of the present invention inhibits cell proliferation of cancer cells by a mechanism dependent on Chkl inhibition.
A solution of tert-butyl 3-(2-(3-(5-bromopyrazin-2-ylamino)-lH-pyrazol-5-yl)-3- methoxyphenoxy)propylcarbamate (0.378 g, 0.730 mmol) and zinc cyanide (0.10 g, 0.870 mmol) in DMF (10 mL) is degassed with a stream of nitrogen for one hour and then -25- heated to 80 0C. To the reaction is added Pd(Ph3P)4 (0.080 g, 0.070 mmol), and the mixture is heated overnight. The reaction is cooled to room temperature and concentrated under reduced pressure. The residue is purified by silica gel chromatography (CH2Cl2/Me0H) to give 0.251 g (73%) of the title compound.
A 5 L flange-neck round-bottom flask equipped with an air stirrer rod and paddle, thermometer, pressure-equalizing dropping funnel, and nitrogen bubbler is charged with 5-(5-(2-hydroxy-6-methoxy-phenyl)-lH-pyrazol-3-ylamino)-pyrazine-2-carbonitrile (47.0 g, 152 mmol) and anhydrous THF (1.2 L). The stirred suspension, under nitrogen, is cooled to 0 0C. A separate 2 L 3 -necked round-bottom flask equipped with a large -28- magnetic stirring bar, thermometer, and nitrogen bubbler is charged with triphenylphosphine (44.0 g; 168 mmol) and anhydrous THF (600 mL). The stirred solution, under nitrogen, is cooled to 0 0C and diisopropylazodicarboxylate (34.2 g; 169 mmol) is added and a milky solution is formed. After 3-4 min, a solution of7-butyl-N-(3- hydroxypropyl)-carbamate (30.3 g, 173 mmol) in anhydrous THF (100 mL) is added and the mixture is stirred for 3-4 min. This mixture is then added over 5 min to the stirred suspension of starting material at 0 0C. The reaction mixture quickly becomes a dark solution and is allowed to slowly warm up to room temperature. After 6.5 h, more reagents are prepared as above using PPh3 (8 g), DIAD (6.2 g) and carbamate (5.4 g) in anhydrous THF (150 mL). The mixture is added to the reaction mixture, cooled to -5 0C and left to warm up to room temperature overnight. The solvent is removed in vacuo. The resulting viscous solution is loaded onto a pad of silica and product is eluted with ethyl acetate. The concentrated fractions are separately triturated with methanol and resulting solids are collected by filtration. The combined solids are triturated again with methanol (400 mL) and then isolated by filtration and dried in vacuo at 50 0C overnight to give 31.3 g of desired product. LC-ES/MS m/z 466.2 [M+ 1]+.
A 5 L flange-neck, round-bottom flask equipped with an air stirrer rod and paddle, thermometer, and air condenser with bubbler attached, is charged with tert-bvXyl 3-(2-(3- (5-cyanopyrazin-2-ylamino)-lH-pyrazol-5-yl)-3-methoxyphenoxy)propylcarbamate (30.9 g, 66.3 mmol) and ethyl acetate (3 L). The mechanically stirred yellow suspension is cooled to just below 10 0C. Then hydrogen chloride from a lecture bottle is bubbled in -29- vigorously through a gas inlet tube for 15 min with the ice-bath still in place. After 5 h the mixture is noticeably thickened in appearance. The solid is collected by filtration, washed with ethyl acetate, and then dried in vacuo at 60 0C overnight to give 30.0 g. 1H NMR (400 MHz, DMSO-d6) δ 2.05 (m, 2H), 2.96 (m, 2H), 3.81 (s, 3H), 4.12 (t, J = 5.8 Hz, 2H), 6.08 (br s, 3H), 6.777 (d, J = 8.2 Hz, IH), 6.782 (d, J = 8.2 Hz, IH), 6.88 (br s, IH), 7.34 (t, J = 8.2 Hz, IH), 8.09 (br s, IH), 8.55 (br s, IH), 8.71 (s, IH), 10.83 (s, IH), 12.43 (br s, IH). LC-ES/MS m/z 366.2 [M+lf.
Example 3 5 -(5 -(2-(3 -Aminopropoxy)-6-methoxyphenyl)- 1 H-pyrazol-3 -ylamino)pyrazine-2- carbonitrile
5-(5-(2-(3-Aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-ylamino)pyrazine-2- carbonitrile dihydrogen chloride salt (3.0 g, 6.84 mmol) is suspended in 200 mL of CH2Cl2. 1 N NaOH is added (200 mL, 200 mmol). The mixture is magnetically stirred under nitrogen at room temperature for 5 h. The solid is collected by filtration and washed thoroughly with water. The filter cake is dried in vacuo at 50 0C overnight to give 2.26 g (90%) of the free base as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 1.81 (m, 2H), 2.73 (t, J = 6.2 Hz, 2H), 3.82 (s, 3H), 4.09 (t, J = 6.2 Hz, 2H), 6.76 (t, J = 8.2 Hz, 2H), 6.93 (br s, IH), 7.31 (t, J = 8.2 Hz, IH), 8.52 (br s, IH), 8.67 (s, IH). LC- MS /ES m/z 366.2 [M+ 1]+.
5-(5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-ylamino)pyrazine-2- carbonitrile (1.0 g, 2.74 mmol) is suspended in MeOH (100 mL). A I M solution of methanesulfonic acid in MeOH (2.74 mL, 2.74 mmol) is added to the mixture dropwise with stirring. The solid nearly completely dissolves and is sonicated and stirred for 15 min, filtered, and concentrated to 50 mL. The solution is cooled overnight at -15 0C and the solid that forms is collected by filtration. The solid is dried in a vacuum oven overnight to give 0.938 g (74%) of a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 1.97 (m, 2H), 2.28 (s, 3H), 2.95 (m, 2H), 3.79 (s, 3H), 4.09 (t, J = 5.9 Hz, 2H), 6.753 (d, J = 8.4 Hz, IH), 6.766 (d, J = 8.4 Hz, IH), 6.85 (br s, IH), 7.33 (t, J = 8.4 Hz, IH), 7.67 (br s, 3H), 8.49 (br s, IH), 8.64 (s, IH), 10.70 (s, IH), 12.31 (s, IH). LC-ES/MS m/z 366.2 [M+l]+.
5-(5-(2-Hydroxy-6-methoxyphenyl)-lH-pyrazol-3-ylamino)pyrazine-2- carbonitrile (618 g, 1.62 mol) is slurried in tetrahydrofuran (6.18 L, 10 volumes) and chilled to -5 to 0 0C with an acetone/ice bath. Triethylamine (330 g, 3.25 mol) is added through an addition funnel over 30 – 40 min at -5 to 5 0C. The resulting slurry is stirred at -5 to 5 0C for 60 – 90 min. The insoluble triethylamine hydrochloride is filtered and the solution of the phenol ((5-(2-hydroxy-6-methoxyphenyl)-lH-pyrazol-3- ylamino)pyrazine-2-carbonitrile) collected in an appropriate reaction vessel. The cake is rinsed with THF (1.24 L). The THF solution of the phenol is held at 15 to 20 0C until needed.
Triphenylphosphine (1074 g, 4.05 mol) is dissolved at room temperature in THF (4.33 L). The clear colorless solution is cooled with an acetone/ice bath to -5 to 5 0C. Diisopropylazodicarboxylate (795 g, 3.89 mol) is added dropwise through an addition funnel over 40 – 60 min, keeping the temperature below 10 0C. The resulting thick white slurry is cooled back to -5 to 0 0C. tert-Butyl 3-hydroxypropylcarbamate (717g, 4.05 moles) is dissolved in a minimum of THF (800 mL). The tert-butyl 3- hydroxypropylcarbamate/THF solution is added, through an addition funnel, over 20 – 30 -35- min at -5 to 5 0C to the reagent slurry. The prepared reagent is stirred in the ice bath at -5 to 0 0C until ready for use.
The prepared reagent slurry (20%) is added to the substrate solution at 15 to 20 0C. The remaining reagent is returned to the ice bath. The substrate solution is stirred at ambient for 30 min, then sampled for HPLC. A second approximately 20% portion of the reagent is added to the substrate, stirred at ambient and sampled as before. Addition of the reagent is continued with monitoring for reaction completion by HPLC. The completed reaction is concentrated and triturated with warm methanol (4.33 L, 50 – 60 0C) followed by cooling in an ice bath. The resulting yellow precipitate is filtered, rinsed with cold MeOH (2 L), and dried to constant weight to provide 544 g (72%) of the title compound, mp 214 – 216 0C; ES/MS m/z 466.2 [M+l]+.
tert-Butyl 3-(2-(3-(5-cyanopyrazin-2-ylamino)-lH-pyrazol-5-yl)-3- methoxyphenoxy)propylcarbamate (1430 g, 3.07 mol) is slurried with acetone (21.5 L) in a 30 L reactor. Methanesulfonic acid (1484 g, 15.36 mol) is added through an addition funnel in a moderate stream. The slurry is warmed to reflux at about 52 0C for 1 to 3 h and monitored for reaction completion by HPLC analysis. The completed reaction is cooled from reflux to 15 to 20 0C over 4.5 h. The yellow slurry of 2-pyrazinecarbonitrile, 5-[[5-[-[2-(3-aminopropyl)-6-methoxyphenyl]-lH-pyrazol-3-yl]amino] dimesylate salt is filtered, rinsed with acetone (7 L) and dried in a vacuum oven. The dimesylate salt, (1608 g, 2.88 mol) is slurried in water (16 L). Sodium hydroxide (aqueous 50%, 228 g, 2.85 mol) is slowly poured into the slurry. The slurry is -36- heated to 60 0C and stirred for one hour. It is then cooled to 16 0C over 4 h and filtered. The wet filter cake is rinsed with acetone (4 L) and dried to constant weight in a vacuum oven at 40 0C to provide 833 g (94%) of 2-pyrazinecarbonitrile, 5-[[5-[-[2-(3- aminopropyl)-6-methoxyphenyl]-lH-pyrazol-3-yl]amino] monomesylate monohydrate. mp 222.6 0C; ES/MS m/z 366.2 [M+l]+.
Crude 2-pyrazinecarbonitrile, 5 -[ [5 – [- [2-(3 -aminopropyl)-6-methoxyphenyl]- IH- pyrazol-3-yl] amino] monomesylate monohydrate is purified using the following procedure. The technical grade 2-pyrazinecarbonitrile, 5-[[5-[-[2-(3-aminopropyl)-6- methoxyphenyl]-lH-pyrazol-3-yl] amino] mono mesylate mono hydrate (1221 g, 2.55 mol) is slurried in a solvent mixture of 1: 1 acetone/water (14.7 L). The solid is dissolved by warming the mixture to 50 – 55 0C. The solution is polish filtrated while at 50 – 55 0C through a 0.22μ cartridge filter. The solution is slowly cooled to the seeding temperature of about 42 – 45 0C and seeded. Slow cooling is continued over the next 30 – 60 min to confirm nucleation. The thin slurry is cooled from 38 to 15 0C over 3 h. A vacuum distillation is set up and the acetone removed at 110 – 90 mm and 20 – 30 0C. The mixture is cooled from 30 to 15 0C over 14 h, held at 15 0C for 2 h, and then filtered. The recrystallized material is rinsed with 19: 1 water/acetone (2 L) and then water (6 L) and dried to constant weight in a vacuum oven at 40 0C to provide 1024 g (83.9%) of the title compound, mp 222.6 0C; ES/MS m/z 366.2 [M+l]+. X-ray powder diffraction (XRPD) patterns may be obtained on a Bruker D8
Advance powder diffractometer, equipped with a CuKa source (λ=l.54056 angstrom) operating at 40 kV and 40 mA with a position-sensitive detector. Each sample is scanned between 4° and 35° in °2Θ ± 0.02 using a step size of 0.026° in 2Θ ± 0.02 and a step time of 0.3 seconds, with a 0.6 mm divergence slit and a 10.39 mm detector slit. Primary and secondary Soller slits are each at 2°; antiscattering slit is 6.17 mm; the air scatter sink is in place. -37-
Characteristic peak positions and relative intensities:
Differential scanning calorimetry (DSC) analyses may be carried out on a Mettler- Toledo DSC unit (Model DSC822e). Samples are heated in closed aluminum pans with pinhole from 25 to 350 0C at 10 °C/min with a nitrogen purge of 50 mL/min. Thermogravimetric analysis (TGA) may be carried out on a Mettler Toledo TGA unit (Model TGA/SDTA 85Ie). Samples are heated in sealed aluminum pans with a pinhole from 25 to 350 0C at 10 0C /min with a nitrogen purge of 50 mL/min.
The thermal profile from DSC shows a weak, broad endotherm form 80 – 1400C followed by a sharp melting endotherm at 222 0C, onset (225 0C, peak). A mass loss of 4% is seen by the TGA from 25 – 140 0C.
Combine l-(2-hydroxy-6-methox>’phenyl)e1han-l-one (79.6 kg, 479 mol) and 1,1-<iimethoxy-N,N-dimemylmethanamino (71.7 kg, 603.54 mol) with DMF (126 kg). Heat to 85-90 °C for 12 hours. Cool the reaction mixture containing intermediate (E)-3-(dimethylamino)-l-(2-hydroxy-6-methoxyphenyl)prop-2-en-l-one (mp 84.74 °C) to ambient temperature and add anhydrous potassium phosphate (136 kg, 637.07 mol) and tert-butyl (3-bromopropyl)carbamate (145 kg, 608.33 mol). Stir the reaction for 15 hours at ambient temperature. Filter the mixture and wash the filter cake with ΜΓΒΕ (3 χ , 433 kg, 300 kg, and 350 kg). Add water (136 kg) and aqueous sodium chloride (25% solution, 552 kg) to the combined MTBE organic solutions. Separate the organic and aqueous phases. Back-extract the resulting aqueous phase with MTBE (309 kg) and add the MTBE layer to the organic solution. Add an aqueous sodium chloride solution (25% solution, 660 kg) to the combined organic extracts and separate the layers. Concentrate the organic extracts to 1,040 kg – 1,200 kg and add water (400 kg) at 30-35 °C to the residue. Cool to ambient temperature and collect material by filtration as a wet cake to give the title product (228.35 kg, 90%). ES/MS (m/z): 379.22275 (M+l).
methoxyphenoxy)propyl)carbamate (228.35 kg, 72% as a wet water solid, 434.9 mol) to form a solution. Heat the solution to 35 – 40 °C for 4-6 hours. Cool the reaction to ambient temperature and concentrate to a residue. Add MTBE (300 kg) to the residue and concentrate the solution to 160 kg – 240 kg. Add MTBE (270 kg) and concentrate the solution. Add MTBE (630 kg), water (358 kg), and sodium chloride solution (80 kg, 25% aqueous) and stir for 20 minutes at ambient temperature. Let the mixture stand for 30 minutes. Separate the aqueous layer. Add water (360 kg) and sodium chloride solution (82 kg, 25% sodium chloride) to the organic phase. Stir for 20 minutes at ambient temperature. Let the mixture stand for 30 minutes. Separate the aqueous portion. Add sodium chloride solution (400 kg, 25 % aqueous) to the organic portion. Stir for 20 minutes at ambient temperature. Let the mixture stand for 30 minutes at ambient temperature. Separate the aqueous portion. Concentrate the organic portion to 160 kg – 240 kg at 40 °C. Add ethanol (296 kg) to the organic portion. Concentrate the solution to 160 kg to 240 kg at 40 °C to provide an intermediate of tert-butyl (3-(2-(isoxazol-5-yl)-3-methox>’phenoxy)propyl)carbamate. Add ethanol (143 kg) and water (160 kg) to the concentrated solution. Add potassium hydroxide (31.8 kg) at 40 °C. Add ethanol (80 kg) and adjust the temperature to 45-50 °C. Stir for 4-6 hours at 45-50 °C and concentrate to 160 kg – 240 kg at 40 °C. Add water to the concentrate (160 kg) and acetic acid (9.0 kg) drop-wise to adjust the pH to 10-12 while mamtaining the temperature of the solution at 25 to 35 °C. Add ethyl acetate (771 kg) and acetic acid drop-wise to adjust the pH to 5-7 while maintaining the temperature of the solution at 25-35 °C. Add sodium chloride solution (118 kg, 25% aqueous solution). Stir the mixture for 20 minutes at ambient temperature. Let the solution stand for 30 minutes at ambient temperature. Separate Ihe aqueous portion. Heat the organic portion to 30-35 °C. Add water (358 kg) drop-wise. Stir the solution for 20 minutes while maintaining the temperature at 30 to 35 °C. Let the mixture stand for 30 minutes and separate the aqueous portion. Wash the organic portion with sodium chloride solution (588 kg, 25% aqueous) and concentrate the organic portion to 400 kg – 480 kg at 40-50 °C. Heat the concentrated solution to 50 °C to form a solution. Maintain the solution at 50 °C and add M-heptane (469 kg) drop-wise. Stir the solution for 3 hours at 50 °C before slowly cooling to ambient temperature to crystallize the product. Stir at ambient temperature for 15 hours and filter the crystals. Wash the crystals with ethanol/«-heptane (1 :2, 250 kg) and dry at 45 °C for 24 hours to provide the title compound (133.4 kg, 79.9%), rap. 104.22 °C,
Combine a THJF solution (22%) of fcrt-butyl (3-(2-(2-cyanoacetyl)-3-memoxyphenoxy)propyl)carbamate (1.0 eqv, this is define as one volume) with hydrazine (35%, 1.5 eqv), acetic acid (glacial, 1.0 eqv), water (1 volume based on the THF solution) and methanol (2 volumes based on the THF solution). This is a continuous operation. Heat the resulting mixture to 130 °C and 1379 kPa with a rate of V/Q = 70 minutes, tau = 60. Extract the solution with toluene (4 volumes), water (1 volume), and sodium carbonate (10% aqueous, 1 eqv). Isolate Ihe toluene layer and add to DMSO (0.5 volumes). Collect a solution of the intermediate compound tert-butyl (3-(2-(3-amino-lH-pyrazol-5-yl)-3-methoxyphenoxy) propyl)carbamate (26.59 kg, 91%) in 10 days, mp = 247.17 °C as a DMSO solution (3 volumes of product). N-Eftylmorpholine (1.2 eqv) and 5-chloropyrazine-2-carbonitrile (1.15 eqv) in 2 volumes of DMSO is combined in a tube reactor at 80 °C, V/Q = 3 and tau = 170 minutes at ambient pressure. Add the product stream to methanol (20 vol). As a continuous process, filter the mixture and wash with methanol followed by MTBE. Air dry the material on the filter to give tert-butyl (3-(2-(3-((5-cyanopyrazm-2-yl)arnino)-lH-pyrazol-5-yl)-3-methox>’phenoxy) propyl)carbamate in a continuous fashion (22.2 kg, 88.7%, 8 days). Dissolve a solution of fcrt-butyl (3-(2-(3-((5-cyanopyrazin-2-yl)amino)-lH-pyrazol-5-yl)-3-methoxyphenoxy) propyl)carbamate in formic acid (99%, 142 kg) at ambient temperature and agitate for 4 hours to provide an intermediate of 5-((5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-yl)amino)pyrazine-2-carbonitrile formate. Dilute the solution with water (55 kg), (S)-lactic acid (30%, 176 kg) and distill the resulting mixture until < 22 kg formic acid remains. Crystallize the resulting residue from THF and wash with a THF -water (0.5% in THF) solution. Dry the wet cake at 30 °C at >10% relative humidity to give the title product as a white to yellow solid (24.04 kg, 85-90%), mp. 157 °C.
Add 5-({3-[2-(3-aminopropoxy)-6-methoxyphenyl]-lH-pyrazol-5-yl}ammo)pyrazine-2-carbonitrile (4.984 g, 13.33 mmol, 97.7 wt%) to n-PrOH (15.41 g, 19.21 mL) to form a slurry. Heat the slurry to 60 °C. Add (S)-lactic acid (1.329 g, 14.75 mmol) to water (19.744 mL) and add this solution to the slurry at 58 °C. Heat the solution to 60 °C and add n-PrOH (21.07 g, 26.27 mL). Seed the solution with 5-((5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-yl)ammo)pyrazme-2-carbom^ (S)-lactate monohydrate (48.8 mg, 0.1 mmol) and cool the solution to 40 °C over 35 minutes. Add H-PrOH (60.5 mL) to the slurry at 40 °C via a syringe pump over 2 hours and maintain the temperature at 40 °C. Once complete, air cool the slurry to ambient temperature for 2 hours, the cool the mixture in ice-water for 2 hours. Filter the product, wash the wet cake with 6:1 (v/v) rc-PrOH : H20 (15 mL), followed by n-PrOH (15 mL) and dry the wet cake for 20 minutes. Dry the solid overnight at 40 °C in vacuo to give the title compound as a white to yellow solid (5.621 g, 89.1%), m.p. 157 °C.
Crystalline Example 1
Crystalline 5-(5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3- ylamino)pyrazine-2-carbonitrile (S)-lactate monohydrate Prepare a slurry having 5-(5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3 -y lamino)py razine-2-carbonitrile (368 mg, 1.0 mmol) in a 10:1 THF-water (5 mL) solution and stir at 55 °C. Add (S)-lactic acid (110 mg, 1.22 mmol) dissolved in THF (1 mL). A clear solution forms. Stir for one hour. Reduce Ihe temperature to 44 °C and stir until an off-white precipitate forms. Filter the material under vacuum, rinse with THF, and air dry to give the title compound (296 mg, 80%).
X-Ray Powder Diffraction, Crystalline Example 1 Obtain the XRPD patterns of the crystalline solids on a Bruker D4 Endeavor X-ray powder diffractometer, equipped with a CuKa source (λ = 1.54060 A) and a Vantec detector, operating at 35 kV and 50 mA. Scan the sample between 4 and 40° in 2Θ, with a step size of 0.0087° in 2Θ and a scan rate of 0.5 seconds/step, and with 0.6 mm divergence, 5.28mm fixed anti-scatter, and 9.5 mm detector slits. Pack the dry powder on a quartz sample holder and obtain a smooth surface using a glass slide. It is well known in the crystallography art that, for any given crystal form, the relative intensities of the diffraction peaks may vary due to preferred orientation resulting from factors such as crystal morphology and habit. Where the effects of preferred orientation are present, peak intensities are altered, but the characteristic peak positions of the polymorph are unchanged. See, e.g. The U. S. Pharmacopeia 35 – National Formulary 30 Chapter <941> Characterization of crystalline and partially crystalline solids by XRPD Official December 1, 2012-May 1, 2013. Furthermore, it is also well known in the
crystallography art that for any given crystal form the angular peak positions may vary slightly. For example, peak positions can shift due to a variation in the temperature or humidity at which a sample is analyzed, sample displacement, or the presence or absence of an internal standard. In the present case, a peak position variability of ± 0.2 in 2Θ will take into account these potential variations without hindering the unequivocal identification of the indicated crystal form Confirmation of a crystal form may be made based on any unique combination of distinguishing peaks (in units of ° 2Θ), typically the more prominent peaks. The crystal form diffraction patterns, collected at ambient temperature and relative humidity, were adjusted based on NIST 675 standard peaks at 8.85 and 26.77 degrees 2-theta,
Characterize a prepared sample of crystalline 5-(5-(2-(3-aminopropoxy)-6-methoxyphenyl)- lH-pyrazol-3-ylamino)pyrazine-2-carbonitrile (S)-lactate monohydrate by an XPRD pattern using CuKa radiation as having diffraction peaks (2-theta values) as described in Table 1 below. Specifically the pattern contains a peak at 12.6 in
combination with one or more of the peaks selected from the group consisting of 24.8, 25.5, 8.1, 6.6, 12.3, and 16.3 with a tolerance for the diffraction angles of 0.2 degrees.
Combine a THF solution (22%) of i<?ri-butyl (3-(2-(2-cyanoacetyl)-3-methoxyphenoxy)propyl)carbamate (1.0 eqv, this is define as one volume) with hydrazine (35%, 1.5 eqv), acetic acid (glacial, 1.0 eqv), water (1 volume based on the THF solution) and methanol (2 volumes based on the THF solution). As this is a continuous operation, grams or kg is irrelevant in this processing methodology. Heat the resulting mixture to 130 °C and 1379 kPa with a rate of V/Q = 70 minutes (where V refers to the volume of the reactor and Q refers to flow rate), tau = 60. Extract the solution with toluene (4 volumes), water (1 volume), and sodium carbonate (10% aqueous, 1 eqv). Isolate the toluene layer and add to DMSO (0.5 volumes). Collect a solution of the intermediate compound i<?ri-butyl (3-(2-(3-amino- lH-pyrazol-5-yl)-3-methoxyphenoxy)
propyl)carbamate (26.59 kg, 91%) in 10 days, mp = 247.17 °C as a DMSO solution (3 volumes of product). N-ethylmorpholine (1.2 eqv) and 5-chloropyrazine-2-carbonitrile (1.15 eqv) in 2 volumes of DMSO is combined in a tube reactor at 80 °C, V/Q = 3 and tau = 170 minutes at ambient pressure. Add the product stream to methanol (20 vol). As a continuous process, filter the mixture and wash with methanol followed by MTBE. Air dry the material on the filter to give i<?ri-butyl (3-(2-(3-((5-cyanopyrazin-2-yl)amino)-lH-pyrazol-5-yl)-3-methoxyphenoxy) propyl)carbamate in a continuous fashion (22.2 kg, 88.7%, 8 days). Dissolve a solution of i<?ri-butyl (3-(2-(3-((5-cyanopyrazin-2-yl)amino)-lH-pyrazol-5-yl)-3-methoxyphenoxy) propyl)carbamate in formic acid (99%, 142 kg) at ambient temperature and agitate for 4 hours to provide an intermediate of 5-((5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-yl)amino)pyrazine-2-carbonitrile formate. Dilute the solution with water (55 kg), (S)-lactic acid (30%, 176 kg) and distill the resulting mixture until < 22 kg formic acid remains. Crystallize the resulting residue from THF and wash with a THF -water (0.5% in THF) solution. Dry the wet cake at 30 °C at >10% relative humidity to give the title product as a white to yellow solid (24.04 kg, 85-90%), m.p. 157 °C.
Add 5-({3-[2-(3-aminopropoxy)-6-methoxyphenyl]-lH-pyrazol-5-yl}amino)pyrazine-2-carbonitrile (4.984 g, 13.33 mmol, 97.7 wt%) to n-PrOH (15.41 g, 19.21 mL) to form a slurry. Heat the slurry to 60 °C. Add (S)-lactic acid (1.329 g, 14.75 mmol) to water (19.744 mL) and add this solution to the slurry at 58 °C. Heat the solution to 60 °C and add n-PrOH (21.07 g, 26.27 mL). Seed the solution with 5-((5-(2-(3-aminopropoxy)-6-methoxyphenyl)-lH-pyrazol-3-yl)amino)pyrazine-2-carbonitrile (S)-lactate monohydrate (48.8 mg, 0.1 mmol) and cool the solution to 40 °C over 35 minutes. Add ft-PrOH (60.5 mL) to the slurry at 40 °C via a syringe pump over 2 hours and maintain the temperature at 40 °C. Once complete, air cool the slurry to ambient temperature for 2 hours, then cool the mixture in ice-water for 2 hours. Filter the product, wash the wet cake with 6:1 (v/v) n-PrOH : H20 (15 mL), followed by n-PrOH (15 mL)
and dry the wet cake for 20 minutes. Dry the solid overnight at 40 °C in vacuo to give the title compound as a white to yellow solid (5.621 g, 89.1%), m.p. 157 °C.
Clip
Kilogram-scale prexasertib monolactate monohydrate synthesis under continuous-flow CGMP conditions
Science 16 Jun 2017:
Vol. 356, Issue 6343, pp. 1144-1150
DOI: 10.1126/science.aan0745
science2017, 356, 1144
Kilogram-Scale Prexasertib Monolactate Monohydrate Synthesis under Continuous-Flow CGMP Conditions
A multidisciplinary team from Eli Lilly reports the development and implementation of eight continuous unit operations for the synthesis of ca. 3 kg API per day under CGMP conditions (K. P. Cole et al., Science2017, 356, 1144). The recent drive toward more potent APIs that have a low annual demand (<100 kg) has made continuous synthesis a viable alternative to traditional batch processes with advantages which include reducing equipment footprint and worker exposure. In this report the authors describe the enablement of three continuous synthetic steps followed by a salt formation, using surge tanks between steps to allow each step to be taken offline if online PAT detects a loss in reaction performance. A combination of MSMPRs (mixed-suspension, mixed-product removal) vessels, plug-flow reactors, and dissolve-off filters were used to perform the chemistry, with an automated 20 L rotary evaporator used to concentrate process streams and perform solvents swaps. This paper gives an excellent account of the potential solutions to continuous API synthesis and is well worth a read for anyone contemplating such methodology.
Integrated flow synthesis and purification process for prexasertib meets high industry standards
Continuous crystallisation, shown here, and subsequent filtration have been the most difficult-to-develop part of the prexasertib production process
Eli Lilly has taken an important step away from traditional batch process drug manufacturing by using an industry-first continuous process to make a compound for phase I and II clinical trials. Workers at Lilly’s Kinsale site in Ireland, did three steps involved in producing cancer drug candidate prexasertib continuously, under current good manufacturing practice (CGMP) standards that ensure safety for human consumption.
Continuous processing relies on chemical and physical changes happening as substances flow through pipes. Isolated steps of this type are already well-established in the pharmaceutical industry. However, Lilly principal research scientist Kevin Cole stresses that a series including reaction and purification steps like this has not been demonstrated before. And the company wants to go much further.
‘We envision entire synthetic routes consisting of many reaction and separation unit operations being executed simultaneously in flow, with heavy reliance on design space understanding, process analytical technologies and process modelling to ensure quality,’ Cole says. ‘We think this will drastically change the environment for pharmaceutical manufacturing.’
The complex synthesis of prexasertib even requires the use of toxic hydrazine – used as a rocket fuel. As a result, and because of prexasertib’s toxicity, the drug was a good candidate to test out a comprehensive flow chemistry setup
In batch processes different chemical reaction and purification steps are typically done in large, costly vessels. However, this can be uneconomical when small amounts of drug molecules are needed for early stage clinical trials and, because drugs are getting more potent, increasingly in mainstream production.
By contrast, small volume continuous flow processing runs in more compact equipment in fume hoods. Flow systems can adapt to different processes, with cheap parts that can either be dedicated to specific drugs or readily replaced. The US Food and Drug Administration (FDA) has also been promoting continuous manufacturing because it integrates well with advanced process analytical technology. This helps pharmaceutical companies make high quality drugs with less FDA oversight.
Lilly chose prexasertib as its test case for such a process because it’s challenging to make. It is a chain of three aromatic rings, and one challenge comes because its central ring is formed using hydrazine. Hydrazine is used as a component in rocket fuel, and is also highly toxic. A second challenge comes from prexasertib itself, which, as a potent kinase inhibitor, is toxic to healthy cells, as well as cancerous ones, even at low doses. Lilly therefore wants to minimise its workers’ exposure.
Feeding the plant
Cole and his colleagues at Lilly’s labs in Indianapolis, US, have developed flow processes for three of the seven steps involved in prexasertib production. They start with the hydrazine step, which they could safely speed up by super-heating in the continuous process. After aqueous workup purification the solution of the two-ring intermediate solution runs into a ‘surge tank’. From there the solution flows intermittently into a rotary evaporator that removes solvents to concentrate it.
The second continuous flow step adds the third of prexasertib’s rings. In this case, the Lilly team purified the intermediate by crystallising it and filtering it out, washing away impurities. They could then redissolve the pure intermediate in formic acid, which also removes a protecting group, giving the desired prexasertib molecule. Automating this was probably the hardest part, Cole says. ‘Development of a predictive filtration model, equipment design and identification of formic acid as the solvent were keys to success,’ he explains. The final flow step then starts converting prexasertib to its final lactate salt form.
This coil of tubes forms a low-cost deprotection gas/liquid reactor Eli Lilly uses during continuous processing of prexasertib
After developing the processes and systems in Indianapolis, Lilly shipped them to be equipped in an existing facility at its Kinsale manufacturing site at the cost of €1 million (£870,000). Once the prexasertib system was installed, the company was able to make 3kg of raw material per day for clinical trials. Cole describes the level of manual intervention needed as ‘moderate’.
Klavs Jensen from the Massachusetts Institute of Technology calls the paper describing the work ‘terrific’. ‘This work marks an important milestone in the continuous manufacturing of pharmaceuticals by demonstrating the feasibility of producing a modern kinase inhibitor under CGMP conditions,’ he says.
Likewise, Brahim Benyahia from Loughborough University, UK, calls this achievement ‘very interesting’. ‘The paper is another example that demonstrates the benefits and feasibility of the integrated continuous approach in pharma,’ he says.
Cole adds that Lilly has several other similar projects in advanced stages of development intended for the €35 million small-volume continuous plant it recently built in Kinsale. ‘We are committed to continuous manufacturing as well as full utilisation of our new facility,’ he says.
Correction: This article was updated on 16 June 2017 to clarify the chronology of the completion of the Kinsale, Ireland plant
1: Lowery CD, VanWye AB, Dowless M, Blosser W, Falcon BL, Stewart J, Stephens J, Beckmann RP, Bence Lin A, Stancato LF. The Checkpoint Kinase 1 Inhibitor Prexasertib Induces Regression of Preclinical Models of Human Neuroblastoma. Clin Cancer Res. 2017 Mar 7. pii: clincanres.2876.2016. doi: 10.1158/1078-0432.CCR-16-2876. [Epub ahead of print] PubMed PMID: 28270495.
2: Zeng L, Beggs RR, Cooper TS, Weaver AN, Yang ES. Combining Chk1/2 inhibition with cetuximab and radiation enhances in vitro and in vivo cytotoxicity in head and neck squamous cell carcinoma. Mol Cancer Ther. 2017 Jan 30. pii: molcanther.0352.2016. doi: 10.1158/1535-7163.MCT-16-0352. [Epub ahead of print] PubMed PMID: 28138028.
3: Ghelli Luserna Di Rorà A, Iacobucci I, Imbrogno E, Papayannidis C, Derenzini E, Ferrari A, Guadagnuolo V, Robustelli V, Parisi S, Sartor C, Abbenante MC, Paolini S, Martinelli G. Prexasertib, a Chk1/Chk2 inhibitor, increases the effectiveness of conventional therapy in B-/T- cell progenitor acute lymphoblastic leukemia. Oncotarget. 2016 Aug 16;7(33):53377-53391. doi: 10.18632/oncotarget.10535. PubMed PMID: 27438145; PubMed Central PMCID: PMC5288194.
REFERENCES
1: Zeng L, Beggs RR, Cooper TS, Weaver AN, Yang ES. Combining Chk1/2 inhibition with cetuximab and radiation enhances in vitro and in vivo cytotoxicity in head and neck squamous cell carcinoma. Mol Cancer Ther. 2017 Jan 30. pii: molcanther.0352.2016. doi: 10.1158/1535-7163.MCT-16-0352. [Epub ahead of print] PubMed PMID: 28138028.
2: Ghelli Luserna Di Rorà A, Iacobucci I, Imbrogno E, Papayannidis C, Derenzini E, Ferrari A, Guadagnuolo V, Robustelli V, Parisi S, Sartor C, Abbenante MC, Paolini S, Martinelli G. Prexasertib, a Chk1/Chk2 inhibitor, increases the effectiveness of conventional therapy in B-/T- cell progenitor acute lymphoblastic leukemia. Oncotarget. 2016 Aug 16;7(33):53377-53391. doi: 10.18632/oncotarget.10535. PubMed PMID: 27438145; PubMed Central PMCID: PMC5288194.
3: King C, Diaz HB, McNeely S, Barnard D, Dempsey J, Blosser W, Beckmann R, Barda D, Marshall MS. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol Cancer Ther. 2015 Sep;14(9):2004-13. doi: 10.1158/1535-7163.MCT-14-1037. PubMed PMID: 26141948.
4: Hong D, Infante J, Janku F, Jones S, Nguyen LM, Burris H, Naing A, Bauer TM, Piha-Paul S, Johnson FM, Kurzrock R, Golden L, Hynes S, Lin J, Lin AB, Bendell J. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J Clin Oncol. 2016 May 20;34(15):1764-71. doi: 10.1200/JCO.2015.64.5788. PubMed PMID: 27044938.
FDA approves new treatment for certain advanced or metastatic breast cancers
The U.S. Food and Drug Administration today approved Verzenio (abemaciclib) to treat adult patients who have hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer that has progressed after taking therapy that alters a patient’s hormones (endocrine therapy). Verzenio is approved to be given in combination with an endocrine therapy, called fulvestrant, after the cancer had grown on endocrine therapy. It is also approved to be given on its own, if patients were previously treated with endocrine therapy and chemotherapy after the cancer had spread (metastasized). Continue reading
The U.S. Food and Drug Administration today approved Verzenio (abemaciclib) to treat adult patients who have hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer that has progressed after taking therapy that alters a patient’s hormones (endocrine therapy). Verzenio is approved to be given in combination with an endocrine therapy, called fulvestrant, after the cancer had grown on endocrine therapy. It is also approved to be given on its own, if patients were previously treated with endocrine therapy and chemotherapy after the cancer had spread (metastasized).
“Verzenio provides a new targeted treatment option for certain patients with breast cancer who are not responding to treatment, and unlike other drugs in the class, it can be given as a stand-alone treatment to patients who were previously treated with endocrine therapy and chemotherapy,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research.
Verzenio works by blocking certain molecules (known as cyclin-dependent kinases 4 and 6), involved in promoting the growth of cancer cells. There are two other drugs in this class that are approved for certain patients with breast cancer, palbociclib approved in February 2015 and ribociclib approved in March 2017.
Breast cancer is the most common form of cancer in the United States. The National Cancer Institute at the National Institutes of Health estimates approximately 252,710 women will be diagnosed with breast cancer this year, and 40,610 will die of the disease. Approximately 72 percent of patients with breast cancer have tumors that are HR-positive and HER2-negative.
The safety and efficacy of Verzenio in combination with fulvestrant were studied in a randomized trial of 669 patients with HR-positive, HER2-negative breast cancer that had progressed after treatment with endocrine therapy and who had not received chemotherapy once the cancer had metastasized. The study measured the length of time tumors did not grow after treatment (progression-free survival). The median progression-free survival for patients taking Verzenio with fulvestrant was 16.4 months compared to 9.3 months for patients taking a placebo with fulvestrant.
The safety and efficacy of Verzenio as a stand-alone treatment were studied in a single-arm trial of 132 patients with HR-positive, HER2-negative breast cancer that had progressed after treatment with endocrine therapy and chemotherapy after the cancer metastasized. The study measured the percent of patients whose tumors completely or partially shrank after treatment (objective response rate). In the study, 19.7 percent of patients taking Verzenio experienced complete or partial shrinkage of their tumors for a median 8.6 months.
Common side effects of Verzenio include diarrhea, low levels of certain white blood cells (neutropenia and leukopenia), nausea, abdominal pain, infections, fatigue, low levels of red blood cells (anemia), decreased appetite, vomiting and headache.
Serious side effects of Verzenio include diarrhea, neutropenia, elevated liver blood tests and blood clots (deep venous thrombosis/pulmonary embolism). Women who are pregnant should not take Verzenio because it may cause harm to a developing fetus.
Ogivri, a biosimilar to the cancer drug Herceptin, is approved for HER2+ breast cancer and metastatic stomach cancers
The U.S. Food and Drug Administration today approved Ogivri (trastuzumab-dkst) as a biosimilar to Herceptin (trastuzumab) for the treatment of patients with breast or metastatic stomach cancer (gastric or gastroesophageal junction adenocarcinoma) whose tumors overexpress the HER2 gene (HER2+). Ogivri is the first biosimilar approved in the U.S. for the treatment of breast cancer or stomach cancer and the second biosimilar approved in the U.S. for the treatment of cancer. Continue reading.
December 1, 2017
Release
The U.S. Food and Drug Administration today approved Ogivri (trastuzumab-dkst) as a biosimilar to Herceptin (trastuzumab) for the treatment of patients with breast or metastatic stomach cancer (gastric or gastroesophageal junction adenocarcinoma) whose tumors overexpress the HER2 gene (HER2+). Ogivri is the first biosimilar approved in the U.S. for the treatment of breast cancer or stomach cancer and the second biosimilar approved in the U.S. for the treatment of cancer.
As with any treatment, health care professionals should review the prescribing information in the labeling for detailed information about the approved uses.
“The FDA continues to grow the number of biosimilar approvals, helping to promote competition that can lower health care costs. This is especially important when it comes to diseases like cancer, that have a high cost burden for patients,” said FDA Commissioner Scott Gottlieb, M.D. “We’re committed to taking new policy steps to advance our biosimilar pathway and promote more competition for biological drugs.”
Biological products are generally derived from a living organism and can come from many sources, such as humans, animals, microorganisms or yeast. A biosimilar is a biological product that is approved based on data showing that it is highly similar to a biological product already approved by the FDA (reference product) and has no clinically meaningful differences in terms of safety, purity and potency (i.e., safety and effectiveness) from the reference product, in addition to meeting other criteria specified by law.
The FDA’s approval of Ogivri is based on review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamic data, clinical immunogenicity data and other clinical safety and effectiveness data that demonstrates Ogivri is biosimilar to Herceptin. Ogivri has been approved as a biosimilar, not as an interchangeable product.
Common expected side effects of Ogivri for the treatment of HER2+ breast cancer include headache, diarrhea, nausea, chills, fever, infection, congestive heart failure, difficulty sleeping (insomnia), cough and rash. Common expected side effects of Ogivri for the treatment of HER2+ metastatic stomach cancer include low levels of certain white blood cells (neutropenia), diarrhea, fatigue, low levels of red blood cells (anemia), inflammation of the mouth (stomatitis), weight loss, upper respiratory tract infections, fever, low levels of blood platelets (thrombocytopenia), swelling of the mucous membranes (mucosal inflammation), common cold (nasopharyngitis) and unusual taste sensation (dysgeusia). Serious expected side effects of Ogivri include worsening of chemotherapy-induced neutropenia.
Like Herceptin, the labeling for Ogivri contains a Boxed Warning to alert health care professionals and patients about increased risks of heart disease (cardiomyopathy), infusions reactions, lung damage (pulmonary toxicity) and harm to a developing fetus (embryo-fetal toxicity). Patients should stop taking Ogivri if cardiomyopathy, life-threatening allergic reactions (anaphylaxis), swelling below the skin (angioedema), inflammation of the lungs (interstitial pneumonitis) or fluid in the lungs (acute respiratory distress syndrome) occur. Patients should be advised of the potential risk to a developing fetus and to use effective contraception.
The FDA granted approval of Ogivri to Mylan GmbH. Herceptin was approved in September 1998 and is manufactured by Genentech, Inc.
/////////////Ogivri, biosimilar , cancer, Herceptin, Trastuzumab, FDA 2017
Biocon Launches KRABEVA® in India, A Biosimilar Bevacizumab for Treating Several Types of Cancer
On November 23, 2017, Biocon India’s premier Biopharmaceuticals Company announced that it has launched KRABEVA®, a biosimilar Bevacizumab for the treatment of patients with metastatic colorectal cancer and other types of lung, kidney, cervical, ovarian and brain cancers, in India 1.
KRABEVA®, a monoclonal antibody (mAb) developed by Biocon, will help expand access to a world-class, high quality biosimilar Bevacizumab for cancer patients in India. It is the world´s first and only Bevacizumab with a unique ´QualCheck ´ mechanism, which ensures that patients get a quality-ascertained product right up to infusion.
Bevacizumab is indicated as a first-line treatment of patients with metastatic colorectal cancer (mCRC), and is accepted as a standard treatment option in combination with chemotherapy for patients with non-small-cell lung cancer (NSLC), metastatic renal cell carcinoma or recurrent ovarian cancer.
KRABEVA® is the second key oncologic biosimilar product, from Biocon´s global biosimilars portfolio to be launched in India. It is being offered to patients at an MRP of Rs 24,000 for 100 mg / 4 ml vials and Rs 39,990 for 400 mg / 16 ml vials, making it a high quality affordable alternative to the innovator brand. In comparison, the Innovator brand for Bevacizumab marketed as Avastin® by Roche India Private Limited costs over Rs 10, 7065 for 400mg / 16ml vial.
Bevacizumab is a monoclonal antibody (mAb) targeting Vascular Endothelial Growth Factor- A (VEGF-A), a cell protein that induces growth of blood vessels that feed tumors. By blocking this protein, Bevacizumab cuts the supply of food and oxygen to the tumor, thus starving it.
Bevacizumab is prescribed in the treatment of several cancers including metastatic colorectal cancer, ovarian cancer, advanced non-small-cell lung cancer, recurrent glioblastoma, cervical cancer and renal cancer. Bevacizumab was first approved by the United States Food and
Drug Administration (USFDA), in February 2004 2.
It also features in the World Health Organization’s (WHO) list of essential medicines 3. The WHO list of essential medicines contains the medications considered to be most effective and safe to meet the most important needs in a health system. The list is frequently used by countries to help develop their own local lists of essential medicine.
Reduction of Chemotherapy-Induced Myelosuppression
Trilaciclib dihydrochloride
1977495-97-8
In phase II clinical development as a chemoprotectant at G1 Therapeutics for first- or second-line treatment in patients with metastatic triple negative breast cancer, in combination with gemcitabine and carboplatin
PATENT, WO 2014144326, Compound 89 (also referred to as Compound T)
Director, Lineberger Comprehensive Cancer Center Founder, G1 Therapeutics ($GTHX)
Notable work
Wellcome Distinguished Professor, American Society of Clinical Investigation Member, Association of American Cancer Institute board of directors,
NCI Director Dr. Norman E. Sharpless
NCI Director Dr. Norman E. Sharpless, Credit: National Institutes of Health
Norman E. “Ned” Sharpless, M.D., was officially sworn in as the 15th director of the National Cancer Institute (NCI) on October 17, 2017. Prior to his appointment, Dr. Sharpless served as the director of the University of North Carolina (UNC) Lineberger Comprehensive Cancer Center, a position he held since January 2014.
Dr. Sharpless was a Morehead Scholar at UNC–Chapel Hill and received his undergraduate degree in mathematics. He went on to pursue his medical degree from the UNC School of Medicine, graduating with honors and distinction in 1993. He then completed his internal medicine residency at the Massachusetts General Hospital and a hematology/oncology fellowship at Dana-Farber/Partners Cancer Care, both of Harvard Medical School in Boston.
After 2 years on the faculty at Harvard Medical School, he joined the faculty of the UNC School of Medicine in the Departments of Medicine and Genetics in 2002. He became the Wellcome Professor of Cancer Research at UNC in 2012.
Dr. Sharpless is a member of the Association of American Physicians as well as the American Society for Clinical Investigation (ASCI), the nation’s oldest honor society for physician–scientists, and served on the ASCI council from 2011 to 2014. Dr. Sharpless was an associate editor of Aging Cell and deputy editor of the Journal of Clinical Investigation. He has authored more than 150 original scientific papers, reviews, and book chapters, and is an inventor on 10 patents. He cofounded two clinical-stage biotechnology companies: G1 Therapeutics and HealthSpan Diagnostics.
In addition to serving as director of NCI, Dr. Sharpless continues his research in understanding the biology of the aging process that promotes the conversion of normal self-renewing cells into dysfunctional cancer cells. Dr. Sharpless has made seminal contributions to the understanding of the relationship between aging and cancer, and in the preclinical development of novel therapeutics for melanoma, lung cancer, and breast cancer.
Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Combination With Etoposide and Carboplatin in Extensive Stage Small Cell Lung Cancer (SCLC)
Active, not recruiting
2
Synthesis
WO 2016040858
Trilaciclib (G1T28)
Trilaciclib is a potential first-in-class short-acting CDK4/6 inhibitor in development to preserve hematopoietic stem cells and enhance immune system function during chemotherapy. Trilaciclib is administered intravenously prior to chemotherapy and has the potential to significantly improve treatment outcomes.
Data from a Phase 1 trial in healthy volunteers were presented at the American Society of Clinical Oncology 2015 Annual Meeting and published in Science Translational Medicine. Trilacicilib has been extensively studied in animals; these preclinical data have been presented at several scientific meetings and published in Molecular Cancer Therapeutics, Science Translational Medicine, and Cancer Discovery.
Trilaciclib is a small molecule, competitive inhibitor of cyclin dependent kinases 4 and 6 (CDK4/6), with potential antineoplastic and chemoprotective activities. Upon intravenous administration, trilaciclib binds to and inhibits the activity of CDK4/6, thereby blocking the phosphorylation of the retinoblastoma protein (Rb) in early G1. This prevents G1/S phase transition, causes cell cycle arrest in the G1 phase, induces apoptosis, and inhibits the proliferation of CDK4/6-overexpressing tumor cells. In patients with CDK4/6-independent tumor cells, G1T28 may protect against multi-lineage chemotherapy-induced myelosuppression (CIM) by transiently and reversibly inducing G1 cell cycle arrest in hematopoietic stem and progenitor cells (HSPCs) and preventing transition to the S phase. This protects all hematopoietic lineages, including red blood cells, platelets, neutrophils and lymphocytes, from the DNA-damaging effects of certain chemotherapeutics and preserves the function of the bone marrow and the immune system. CDKs are serine/threonine kinases involved in the regulation of the cell cycle and may be overexpressed in certain cancer cell types. HSPCs are dependent upon CDK4/6 for proliferation.
Trilaciclib (G1T28) is a CDK4/6 inhibitor in phase II clinical development as a chemoprotectant at G1 Therapeutics for first- or second-line treatment in patients with metastatic triple negative breast cancer, in combination with gemcitabine and carboplatin. Also, phase II trials are ongoing in newly diagnosed, treatment-naive small-cell lung cancer patients, in combination with carboplatin, etoposide, and atezolizumab and phase I trials in previously treated small-cell lung cancer patients, in combination with topotecan.
U.S. Patent Nos. 8,822,683; 8,598,197; 8,598,186, 8,691,830, 8,829,102, 8,822,683, 9, 102,682, 9,499,564, 9,481,591, and 9,260,442, filed by Tavares and Strum and assigned to Gl Therapeutics describe a class of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amine cyclin dependent kinase inhibitors including those of the formula with variables as defined therein):
U.S. Patent Nos. 9,464,092, 9,487,530, and 9,527,857 which are also assigned to Gl Therapeutics describe the use of the above pyrimidine-based agents in the treatment of cancer.
These patents provide a general synthesis of the compounds that is based on a coupling reaction of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine. Such coupling reactions are sometimes referred to as Buchwald coupling (see WO Ί56 paragraph 127; reference WO 2010/020675). The lactam of the fused chloropyrimidine, for example, a 2-chloro-spirocyclo-pyrrolo[2,3-d]pyrimidine-one such as Intermediate K as shown below can be prepared by dehydration of the corresponding carboxylic acid. The reported process to prepare intermediate IK requires seven steps.
(Intermediate IK; page 60, paragraph 215 of WO Ί56)
WO 2013/148748 (U.S. S.N. 61/617,657) entitled “Lactam Kinase Inhibitors” filed by Tavares, and also assigned to Gl Therapeutics likewise describes the synthesis of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines via the coupling reaction of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine.
WO 2013/163239 (U.S. S.N. 61/638,491) “Synthesis of Lactams” describes a method for the synthesis of this class of compounds with the variation that in the lactam preparation step, a carboxylic acid can be cyclized with a protected amine in the presence of a strong acid and a dehydrating agent, which can be together in one moiety as a strong acid anhydride. The purported improvement is that cyclization can occur without losing the protecting group on the amine before cyclization. The typical leaving group is “tBOC” (t-butoxycarbonyl). The application teaches (page 2 of WO 2013/163239) that the strong acid is, for example, trifluoroacetic acid anhydride, tribromoacetic acid anhydride, trichloroacetic acid anhydride or mixed anhydrides. An additional step may be necessary to take off the N-protecting group. The dehydrating agent can be a carbodiimide-based compound such as DCC (Ν,Ν-dicyclohexylcarbodiimide), EDC (l-ethyl-3-(3-dimethylaminopropyl)carbodiimide, or DIC (Ν,Ν-diisopropylcarbodiimide). DCC and DIC are in the same class of reagents-carbodiimides. DIC is sometimes considered better because it is a liquid at room temperature, which facilitates reactions.
WO 2015/061407 filed by Tavares and licensed to Gl Therapeutics also describes the synthesis of these compounds via the coupling of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine. WO ‘407 focuses on the lactam production step and in particular describes that the fused lactams of these compounds can be prepared by treating the carboxylic acid with an acid and a dehydrating agent in a manner that a leaving group on the amine is not removed during the amide-forming ring closing step.
Other publications that describe compounds of this general class include the following. WO 2014/144326 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for protection of normal cells during chemotherapy using pyrimidine based CDK4/6 inhibitors. WO 2014/144596 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for protection of hematopoietic stem and progenitor cells against ionizing radiation using pyrimidine based CDK4/6 inhibitors. WO 2014/144847 filed by Strum et al. and assigned to Gl Therapeutics describes HSPC-sparing treatments of abnormal cellular proliferation using pyrimidine based CDK4/6 inhibitors. WO2014/144740 filed by Strum et al. and assigned to Gl Therapeutics describes highly active anti -neoplastic and anti-proliferative pyrimidine based CDK 4/6 inhibitors. WO 2015/161285 filed by Strum et al. and assigned to Gl Therapeutics describes tricyclic pyrimidine based CDK inhibitors for use in radioprotection. WO 2015/161287 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for the protection of cells during chemotherapy. WO 2015/161283 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for use in HSPC-sparing treatments of RB-positive abnormal cellular proliferation. WO 2015/161288 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for use as anti -neoplastic and anti-proliferative agents. WO 2016/040858 filed by Strum et al. and assigned to Gl Therapeutics describes the use of combinations of pyrimidine based CDK4/6 inhibitors with other anti-neoplastic agents. WO 2016/040848 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for treating certain Rb-negative cancers with CDK4/6 inhibitors and topoisomerase inhibitors.
Other biologically active fused spirolactams and their syntheses are described, for example, in the following publications. Griffith, D. A., et al. (2013). “Spirolactam-Based Acetyl-CoA Carboxylase Inhibitors: Toward Improved Metabolic Stability of a Chromanone Lead Structure.” Journal of Medicinal Chemistry 56(17): 7110-7119, describes metabolically stable spirolactams wherein the lactam resides on the fused ring for the inhibition of acetyl-CoA carboxylase. WO 2013/169574 filed by Bell et al. describes aliphatic spirolactams as CGRP receptor antagonists wherein the lactam resides on the spiro ring. WO 2007/061677 filed by Bell et al. describes aryl spirolactams as CGRP receptor antagonists wherein the lactam resides on the spiro ring. WO 2008/073251 filed by Bell et al. describes constrained spirolactam compounds wherein the lactam resides on the spiro ring as CGRP receptor antagonists. WO 2006/031606 filed by Bell et al. describes carboxamide spirolactam compounds wherein the spirolactam resides on the spiro ring as CGRP receptor antagonists. WO 2006/031610, WO 2006/031491, and WO 2006/029153 filed by Bell et al. describe anilide spirolactam compounds wherein the spirolactam resides on the spiro ring; WO 2008/109464 filed by Bhunai et al. describes spirolactam compounds wherein the lactam resides on the spiro ring which is optionally further fused.
Given the therapeutic activity of selected N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines, it would be useful to have additional methods for their preparation. It would also be useful to have new intermediates that can be used to prepare this class of compounds.
Intermediates B, E, K, L, 1A, IF and 1CA were synthesized according to US 8,598,186 entitled CDK Inhibitors to Tavares, F.X. and Strum, J.C..
The patents WO 2013/148748 entitled Lactam Kinase Inhibitors to Tavares, F.X., WO 2013/163239 entitled Synthesis of Lactams to Tavares, F.X., and US 8,598,186 entitled CDK Inhibitors to Tavares, F.X. and Strum, J.C. are incorporated by reference herein in their entirety. Example 1
Synthesis of tert-butyl N- [2- [(5-bromo-2-chloro-pyrimidin-4yl)amino] ethyl] carbamate, Compound 1
To a solution of 5-bromo-2,4-dichloropyrimidine (3.2 g, 0.0135 mol) in ethanol (80 mL) was added Hunig’s base (3.0 mL) followed by the addition of a solution of N-(tert- butoxycarbonyl)-l,2-diaminoethane (2.5 g, 0.0156 mole) in ethanol (20 mL). The contents were stirred overnight for 20 hrs. The solvent was evaporated under vacuum. Ethyl acetate (200 mL) and water (100 mL) were added and the layers separated. The organic layer was dried with magnesium sulfate and then concentrated under vacuum. Column chromatography on silica gel using hexane/ethyl acetate (0- 60%) afforded tert-butyl N-[2-[(5-bromo-2-chloro-pyrimidin-4- yl)amino]ethyl]carbamate. 1HNMR (d6-DMSO) δ ppm 8.21 (s, 1H), 7.62 (brs, 1H), 7.27 (brs, 1H), 3.39 (m, 2H), 3.12 (m, 2H), 1.34 (s, 9H). LCMS (ESI) 351 (M + H).
Example 2
Synthesis of tert-butyl N-[2-[[2-chloro-5-(3,3-diethoxyprop-l-ynyl)pyrimidin-4- yl] amino] ethyl] carbamate, Compound 2
To tert-butyl N-[2-[(5-bromo-2-chloro-pyrimidin-4-yl)amino]ethyl]carbamate (1.265 g, 6 mmol) in THF (10 mL) was added the acetal (0.778 mL, 5.43 mmol), Pd(dppf)CH2Cl2 (148 g), and triethylamine (0.757 mL, 5.43 mmol). The contents were degassed and then purged with nitrogen. To this was then added Cul (29 mg). The reaction mixture was heated at reflux for 48 hrs. After cooling, the contents were filtered over CELITE and concentrated. Column chromatography of the resulting residue using hexane/ethyl acetate (0- 30%) afforded tert-butyl N- [2- [ [2-chloro-5 -(3 ,3 -diethoxyprop- 1 -ynyl)pyrimidin-4-yl]amino] ethyl] carbamate. 1HNMR (d6-DMSO) δ ppm 8.18 (s, 1H), 7.63 (brs, 1H), 7.40 (brs, 1H), 5.55 (s, 1H), 3.70 (m, 2H), 3.60 (m, 2H), 3.42 (m, 2H), 3.15 (m, 2H), 1.19 – 1.16 (m, 15H). LCMS (ESI) 399 (M + H).
Example 3
Synthesis of tert-butyl N-[2-[2-chloro-6-(diethoxymethyl)pyrrolo[2,3-d]pyrimidin-7- yl] ethyl] carbamate, Compound 3
To a solution of the coupled product (2.1 g, 0.00526 mole) in THF (30 mL) was added TBAF solid (7.0 g). The contents were heated to and maintained at 65 degrees for 2 hrs. Concentration followed by column chromatography using ethyl acetate/hexane (0-50%) afforded tert-butyl N-[2-[2-chloro-6-(diethoxymethyl)pyrrolo[2,3-d]pyrimidin-7-yl]ethyl]carbamate as a pale brown liquid (1.1 g). 1FiNMR (d6-DMSO) δ ppm 8.88 (s, 1H), 6.95 (brs, 1H), 6.69 (s, 1H), 5.79 (s, 1H), 4.29 (m, 2H), 3.59 (m, 4H), 3.34 (m, 1H), 3.18 (m, 1H), 1.19 (m, 9H), 1.17 (m, 6H). LCMS (ESI) 399 (M + H).
Example 4
Synthesis of tert-buty\ N-[2-(2-chloro-6-formyl-pyrrolo [2,3-d] pyrimidin-7- yl)ethyl] carbamate, Compound 4
To the acetal (900 mg) from the preceeding step was added AcOH (8.0 mL) and water
(1.0 mL). The reaction was stirred at room temperature for 16 hrs. Cone, and column chromatography over silica gel using ethyl acetate/hexanes (0- 60%) afforded tert-butyl N-[2-(2- chloro-6-formyl-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl]carbamate as a foam (0.510 g). 1HNMR (d6-DMSO) δ ppm 9.98 (s, 1H), 9.18 (s, 1H), 7.66 (s, 1H), 6.80 (brs, 1H), 4.52 (m, 2H), 4.36 (m, 2H), 1.14 (s, 9H). LCMS (ESI) 325 (M + H).
To the aldehyde (0.940 g) from the preceeding step in DMF (4 mL) was added oxone (1.95 g, 1.1 eq). The contents were stirred at room temp for 7 hrs. Silica gel column chromatography using hexane/ethyl acetate (0- 100%) afforded l-\2-(tert- butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid (0.545 g). 1HNMR (d6-DMSO) δ ppm 9.11 (s, 1H), 7.39 (s, 1H), 4.38 (m, 2H), 4.15 (m, 2H), 1.48 (m, 9H). LCMS (ESI) 341(M + H).
Example 6
Synthesis of methyl 7-[2-(teri-butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3- d]pyrimidine-6-carboxylate, Compound 6
To a solution of 2-chloro-7-propyl-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid (0.545 g, 0.00156 mole) from the preceeding step in toluene (3.5 mL) and MeOH (1 mL) was added TMS- diazomethane (1.2 mL). After stirring overnight at room temperature, the excess of TMS- diazomethane was quenched with acetic acid (3 mL) and the reaction was concentrated under vacuum. The residue was purified by silica gel column chromatography with hexane/ethyl acetate (0- 70%) to afford methyl 7-[2-(tert-butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3- d]pyrimidine-6-carboxylate as an off white solid (0.52 g). 1HNMR (d6-DMSO) δ ppm 9.10 (s, 1H), 7.45 (s, 1H), 6.81 (brs, 1H) 4.60 (m, 2H), 3.91 (s, 3H), 3.29 (m, 2H), 1.18 (m, 9H) LCMS (ESI) 355 (M + H).
Example 7
Synthesis of Chloro tricyclic amide, Compound 7
To methyl 7- [2-(tert-butoxycarbonylamino)ethyl] -2-chloro-pyrrolo [2,3 -d]pyrimidine-6- carboxylate (0.50 g, 0.0014 mole) from the preceeding step in dichloromethane (2.0 mL) was added TFA (0.830 mL). The contents were stirred at room temperature for 1 hr. Concentration under vacuum afforded the crude amino ester which was suspended in toluene (5 mL) and Hunig’s base (0.5 mL). The contents were heated at reflux for 2 hrs. Concentration followed by silica gel column chromatography using hexane/ethyl acetate (0- 50%) afforded the desired chloro tricyclic amide (0.260 g). 1HNMR (d6-DMSO) δ ppm 9.08 (s, 1H), 8.48 (brs, 1H), 7.21 (s, 1H) 4.33 (m, 2H), 3.64 (m, 2H). LCMS (ESI) 223 (M + H).
Example 8
Synthesis of chloro-N-methyltricyclic amide, Compound 8
To a solution of the chloro tricycliclactam, Compound 7, (185 mg, 0.00083 mole) in DMF (2.0 mL) was added sodium hydride (55% dispersion in oil, 52 mg). After stirring for 15 mins, methyl iodide (62 μί, 1.2 eq). The contents were stirred at room temperature for 30 mins. After the addition of methanol (5 mL), sat NaHCOs was added followed by the addition of ethyl acetate. Separation of the organic layer followed by drying with magnesium sulfate and concentration under vacuum afforded the N-methylated amide in quantitative yield. 1FiNMR (d6-DMSO) δ ppm 9.05 (s, 1H), 7.17 (s, 1H) 4.38 (m, 2H), 3.80 (m, 2H), 3.05 (s, 3H). LCMS (ESI) 237 (M + H). Example 9
Synthesis of l-methyl-4-(6-nitro-3-pyridyl)piperazine, Compound 9
To 5-bromo-2-nitropyridine (4.93 g, 24.3 mmole) in DMF (20 mL) was added N- methylpiperazine (2.96 g, 1.1 eq) followed by the addition of DIPEA (4.65 mL, 26.7 mmole). The contents were heated at 90 degrees for 24 hrs. After addition of ethyl acetate (200 mL), water (100 mL) was added and the layers separated. Drying followed by concentration afforded the crude product which was purified by silica gel column chromatography using (0-10%) DCM/Methanol. 1HNMR (d6-DMSO) δ ppm 8.26 (s, 1H), 8.15 (1H, d, J = 9.3 Hz), 7.49 (1H, d, J = 9.4 Hz), 3.50 (m, 4H), 2.49 (m, 4H), 2.22 (s, 3H).
Example 10
Synthesis of 5-(4-methylpiperazin-l-yl)pyridin-2-amine, Compound 10
To l-methyl-4-(6-nitro-3-pyridyl)piperazine (3.4 g) in ethyl acetate (100 mL) and ethanol (100 mL) was added 10%> Pd/C (400 mg) and then the reaction was stirred under hydrogen (10 psi) overnight. After filtration through CELITE, the solvents were evaporated and the crude product was purified by silica gel column chromatography using DCM/ 7N ammonia in MeOH (0- 5%) to afford 5-(4-methylpiperazin-l-yl)pyridin-2-amine (2.2 g). 1HNMR (d6-DMSO) δ ppm 7.56 (1H, d, J = 3 Hz), 7.13 (1H, m), 6.36 (1H, d, J = 8.8 Hz), 5.33 (brs, 2H), 2.88 (m, 4H), 2.47 (m, 4H), 2.16 (s, 3H).
Example 11
Synthesis of tert-butyl 4-(6-amino-3-pyridyl)piperazine-l-carboxylate, Compound 11
This compound was prepared as described in WO 2010/020675 Al .
Synthesis of Compound 89 (also referred to as Compound T)
Compound 89 was synthesized in a similar manner to that described for compound 78 and was converted to an HCl salt. 1HNMR (600 MHz, DMSO-d6) δ ppm 1.47 (br. s., 6 H) 1.72 (br. s., 2 H) 1.92 (br. s., 2 H) 2.77 (br. s., 3 H) 3.18 (br. s., 2 H) 3.46 (br. s., 2 H) 3.63 (br. s., 2 H) 3.66 (d, J=6.15 Hz, 2 H) 3.80 (br. s., 2 H) 7.25 (s, 1 H) 7.63 (br. s., 2 H) 7.94 (br. s., 1 H) 8.10 (br. s., 1 H) 8.39 (br. s., 1 H) 9.08 (br. s., 1 H) 11.59 (br. s., 1 H). LCMS (ESI) 447 (M + H)
The disclosed compounds can be made by the following general schemes:
Scheme 1
In Scheme 1, Ref-1 is WO 2010/020675 Al; Ref-2 is White, J. D.; et al. J. Org. Chem. 1995, 60, 3600; and Ref-3 Presser, A. and Hufher, A. Monatshefte fir Chemie 2004, 135, 1015.
Scheme 2
In Scheme 2, Ref-1 is WO 2010/020675 Al; Ref-4 is WO 2005/040166 Al; and Ref-5 is Schoenauer, K and Zbiral, E. Tetrahedron Letters 1983, 24, 573.
92
93
3) Pd/C/H2
Scheme 6
Scheme 7
NHfOH
Scheme 8
In Scheme 8, Ref-1 is WO 2010/020675 Al; Ref-2 is WO 2005/040166 Al; and Ref-3 is Schoenauer, K and Zbiral, E. Tetrahedron Letters 1983, 24, 573.
Alternatively, the lactam can be generated by reacting the carboxylic acid with a protected amine in the presence of a strong acid and a dehydrating agent, which can be together in one moiety as a strong acid anhydride. Examples of strong acid anhydrides include, but are not limited to, trifluoroacetic acid anhydride, tribromoacetic acid anhydride, trichloroacetic acid anhydride, or mixed anhydrides. The dehydrating agent can be a carbodiimide based compound such as but not limited to DCC (Ν,Ν-dicyclohexylcarbodiimide), EDC (l-ethyl-3-(3-
dimethylaminopropyl)carbodiimide or DIC (Ν,Ν-diisopropylcarbodiimide). An additional step may be necessary to take off the N-protecting group and the methodologies are known to those skilled in the art.
Alternatively, the halogen moiety bonded to the pyrimidine ring can be substituted with any leaving group that can be displaced by a primary amine, for example to create an intermediate for a final product such as Br, I, F, SMe, SO2Me, SOalkyl, SO2alkyl. See, for Exmaple PCT /US2013/037878 to Tavares.
Other amine intermediates and final amine compounds can be synthesized by those skilled in the art. It will be appreciated that the chemistry can employ reagents that comprise reactive functionalities that can be protected and de-protected and will be known to those skilled in the art at the time of the invention. See for example, Greene, T.W. and Wuts, P.G.M., Greene’s Protective Groups in Organic Synthesis, 4th edition, John Wiley and Sons.
Scheme 9
CDK4/6 Inhibitors of the present invention can be synthesized according to the generalized Scheme 9. Specific synthesis and characterization of the Substituted 2-aminopyrmidines can be found in, for instance, WO2012/061156.
Compounds T, Q, GG, and U were prepared as above and were characterized by mass spectrometry and NMR as shown below:
Synthesis of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines. The application appears to be particularly focused on methods for the preparation of trilaciclib and an analog of it. Trilaciclib is the company’s lead CDK4/6 inhibitor presently in phase II trials against small-cell lung cancer and triple negative breast cancer. Interestingly, the company is working on a second CDK4/6 inhibitor, G1T38 , which is in a phase II trial against breast cancer.
GENERAL METHODS
The structure of starting materials, intermediates, and final products was confirmed by standard analytical techniques, including NMR spectroscopy and mass spectrometry. Unless otherwise noted, reagents and solvents were used as received from commercial suppliers. Proton nuclear magnetic resonance spectra were obtained on a Bruker AVANCE 500 at 500 MHz in DMSO-dis. HPLC analyses were performed on a Waters HPLC using the below HPLC method.
HPLC Method
Column: Atlantis T3 (150 χ 4.6, 3 μιη)
Column Temperature: 40°C
Flow Rate: 1 mL/min
Detection: UV @ 275 nm
Analysis Time: 36 min
Mobile Phase A: Water (with 0.1% Trifluoroacetic Acid)
Mobile Phase B : Acetonitrile (with 0.1% Trifluoroacetic Acid)
Sample preparation: dissolve PC sample, wet or dry solid (~1 mg of active compound) in acetonitrile/water (1/1) to achieve complete dissolution.
HPLC Method Gradient
Example 1. General Routes of Synthesis
Scheme 1-1 : Starting from an appropriately substituted halo pyrimidine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the appropriately substituted spirolactam is protected with a group selected from R2. In Step 3 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 3, Step 4, Step 5, or Step 6. Oxidation prior to Step 3 results in undesired byproducts. In Step 4 the hydroxyl group of the fused spirolactam is converted to a leaving group.
In Step 5 the leaving group is dehydrated to afford a compound of Formula IV. In Step 6 the compound of Formula IV is optionally deprotected.
Scheme 1-2: Starting from an appropriately substituted halo pyrimidine compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the appropriately substituted spirolactam is protected with a group selected from R2. In Step 3 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam of Formula IV. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 3 or Step 4. Oxidation prior to Step 3 results in undesired byproducts. In Step 4 the compound of Formula IV is optionally deprotected.
Scheme 1-3 : Starting from an appropriately substituted alkyl glycinate, compounds of the present invention can be prepared. In Step 1 the appropriately substituted alkyl glycinate is subjected to cyclohexanone and TMSCN in the presence of base to afford a cyano species. In Step 2 the appropriately substituted cyanospecies is reduced and subsequently cyclized to afford a compound of Formula I.
Scheme 1-4
Scheme 1-4: Starting from an appropriately substituted l-(aminomethyl)cyclohexan-l-amine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted l-(aminomethyl)cyclohexan-l -amine is reductively aminated with an aldehyde. In Step 2 the appropriately substituted cyclohexane amine is optionally deprotected (i.e.: the group selected from R2 if not H is optionally replaced by H). In Step 3 the cyclohexane amine is cyclized to afford a compound of Formula I. In Step 4 the compound of Formula I is optionally protected.
1-5
Conversion
Scheme 1-5: Starting from an appropriately substituted halo pyrimidine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a
substituted spirolactam. In Step 2 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 2, Step 3, Step 4, or Step 5. Oxidation prior to Step 2 results in undesired byproducts. In Step 3 the hydroxyl group of the fused spirolactam is converted to a leaving group. In Step 4 the leaving group is dehydrated to afford a compound of Formula IV. In Step 5 the compound of Formula IV is optionally deprotected.
S
Scheme 1-6: Starting from an appropriately substituted halo pyrimidine compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam of Formula IV. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 2 or Step 3. Oxidation prior to Step 2 results in undesired byproducts. In Step 3 the compound of Formula IV is optionally deprotected.
Scheme 1-7: Starting from compound of Formula IV a CDK4/6 inhibitor can be prepared. In Step 1 a heteroaryl amine is subjected to a base and a compound of Formula IV is added slowly under chilled conditions to afford a nucleophilic substitution reaction. The compound of Formula IV can previously be prepared as described in the schemes herein.
Example 2. Representative Routes of Synthesis
Scheme 2-1
quant, yield 2 steps
isolated
70% yield 2 steps 75% yield 95% yield
isolated isolated isolated
Scheme 2-1 : An ester route is one embodiment, of the present invention. Ideally, the best synthesis scheme would afford crystalline intermediates to provide material of consistent purity without column chromatography, and high yielding steps while using safe and cost effective reagents when possible.
The first step in the ester route is a SNAr nucleophilic substitution of CI group in commercially available ester 3 using spirolactam 4. Due to low reactivity of 4, a reaction temperature of 85-95 °C was required. Because of the temperature requirements, DIPEA and dimethylacetamide were selected as the base and solvent, respectively. The reaction follows second-order kinetics and usually stalls after -85% conversion. Therefore, the reaction was typically stopped after 60 hours by first cooling it to room temperature at which point solid formation was observed. The mixture was then partitioned between MTBE and water and product was filtered with excellent purity with -53% yield of the desired product 5. The obtained
compound 5 was protected with a Boc group using Boc anhydride and DMAP as the catalyst and dichloromethane as the solvent. The intermediate 6 was obtained in a quantitative yield. Due to the semi-solid nature of compound 6, the material was taken to the next step without further purification. The Dieckmann condensation was initially performed with strong bases such as LiHMDS and tBuOK. A similar result to the aldehyde route (Scheme 2-2) was obtained: a partial deprotection of Boc group was observed that required column chromatography. However, the best results were obtained when DBU was used as base and THF as solvent. The reaction outcome was complete, clean conversion of 6 to 7. Moreover, the product crystallized from the reaction mixture upon seeding, and a quantitative yield was obtained for the two steps.
The hydroxyl group of 7 was removed via a two-step procedure. First, compound 7 was converted completely into triflate 8 using triflic anhydride and triethylamine in dichloromethane. The reaction was found to proceed well at 0°C. Due to the potential instability of the triflate intermediate, it was not isolated. It was immediately taken to the next step and reduced with triethylsilane and palladium tetrakis to afford the product 9 after ethyl acetate crystallization in -70% yield. The Boc group of 9 was removed using trifluoroacetic acid in dichloromethane to afford 10. Intermediate 10 was converted into the final sulfone 11 using Oxone in acetonitrile/water solvent system.
The obtained sulfone 11 was use-tested in the coupling step and was found to perform well. In conclusion, the route to sulfone 11 was developed which eliminated the use of column chromatography with good to excellent yields on all steps.
Scheme 2-2
Molecular Weight: 421
Scheme 2-2: The first step of Scheme 2-2 consistently afforded product 13 contaminated with one major impurity found in substantial amount. Thorough evaluation of the reaction impurity profile by LC-MS and 2D MR was performed, which showed the impurity was structurally the condensation of two aldehyde 12 molecules and one molecule of lactam 4. Therefore, column chromatography was required to purify compound 13, which consistently resulted in a modest 30% yield. A solvent screen revealed that sec-butanol, amyl alcohol, dioxane, and tert-butanol can all be used in the reaction but a similar conversion was observed in each case. However, tert-butanol provided the cleanest reaction profile, so it was selected as a solvent for the reaction. Assessing the impact of varying the stoichiometric ratio of 4 and 12 on the reaction outcome was also investigated. The reaction was performed with 4 equivalents of amine 4 in an attempt to disrupt the 2: 1 aldehyde/amine composition of the impurity. The result was only a marginal increase in product 13 formation. The temperature impact on the reaction outcome was evaluated next. The coupling of aldehyde 12 and 4 was investigated at two different temperatures: 50 °C and 40 °C with 1 : 1 ratio of aldehyde/amine. Reactions were checked at 2 and 4 hours and then every 12 hours. The reaction progress was slow at 50°C and was accompanied by growth of other impurities. The reaction at 40°C was much cleaner; however the conversion was lower in the same time period. The mode of addition of the reagents was investigated as well at 80°C with a slow addition (over 6 hours) of either aldehyde 12 or amine 4 to the reaction mixture. The product distribution did not change and an about 1 to 1 ratio was observed between product and impurity when amine 4 was added slowly to the reaction mixture containing aldehyde 12 and
DIPEA at reflux. The product distribution did change when aldehyde 12 was added slowly to the mixture of amine 4 and DIPEA. However, the major product of the reaction was the undesired impurity. Other organic bases were tried as well as different ratios of DIPEA. No product was observed when potassium carbonate was used as a base. The results of the experiments are presented in Table 1 below.
Table 1
Compound 13 was successfully formed in three cases: triethylamine, 2,6-lutidine and DIPEA, with the DIPEA result being the best. The use of Boc protected spirolactam 4 had no effect on the impurity formation as well. Its utilization was speculated to be beneficial in performing the coupling step together with the following step, preparation of compound 14.
The major impurity formed during Step 1 of Scheme 2-2 is:
Chemical Formula:€2)Η;Μ(¾ 6( 2ί>2
Molecular Weight: 527.4903
The second step (Boc protection of the free lactam) proceeded well using DMAP as a catalyst in dichloromethane at room temperature. The product 14 is a thick oil, and, therefore, cannot be purified by crystallization. The Boc protected intermediate 14 was cyclized successfully into the desired pentacyclic structure 10 upon treatment with a strong base such as LiHMDS or tBuOK. Surprisingly, the Boc group was partially removed during the reaction. The level of deprotection was independent from the internal reaction temperature and was positively correlated with excess of base used. Therefore the mixture of the desired product 10 and 10-Boc compound was treated with acid to completely deprotect Boc group. The conversion of methyl sulfide into the final sulfone 11 was carried out with Oxone. Initially a mixture of methanol and water was used for the reaction. As the result, a partial displacement of sulfone by methoxy group was detected. The methanol was replaced with acetonitrile and the sulfone displacement was eliminated.
In summary, the ester route (Scheme 2-1) is preferred because:
1. Formation of the impurity during the first step of Scheme 2-2 was unavoidable and resulted in yields of < 35%.
2. Column purification was required to isolate intermediate 14.
3. The aldehyde starting material was not commercially available and required two synthetic steps from the corresponding ester.
Scheme 2-3 : Starting with cyclohexanone, compounds of the present invention can be prepared. In Step 1 the methyl glycinate is subjected to cyclohexanone and TMSCN in the presence of tri ethyl amine in DCM to afford 15. In Step 2 15 hydrogenated with hydrogen gas in the presence of catalytic platinum oxide and subsequently undergoes an intramolecular cyclization to afford compound 16 which is used in the schemes above.
Scheme 2-4: Starting with compound 17, compounds of the present invention can be prepared. In Step 1 compound 17 is subjected to ethyl 2-oxoacetate in the presence platinum on carbon and hydrogen gas to afford compound 18. In Step 2 compound 18 is Boc-deprotected with hydrochloric acid. In Step 3 compound 18 is cyclized to afford compound 16 which is used in the schemes above.
Scheme 2-5
11 19
Scheme 2-5: Starting from compound 11 the CDK 4/6 inhibitor 19 can be prepared. In Step 1 5-(4-methylpiperazin-l-yl)pyridin-2-amine is subjected to LiHMDS and compound 11 is added slowly under chilled conditions to afford a nucleophilic substitution reaction and compound 19. Compound 11 can be prepared as described in the schemes herein.
Scheme 2-6: Starting from compound 11 the CDK 4/6 inhibitor 20 can be prepared. In Step 1 5-(4-isopropylpiperazin-l-yl)pyridin-2-amine is subjected to LiHMDS and compound 11 is added slowly under chilled conditions to afford a nucleophilic substitution reaction and compound 20. Compound 11 can be prepared as described in the schemes herein.
Preparation of Compound 5:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet, and reflux condenser was charged with ethyl 4-chloro-2-(methylthio)pyrimidine-5-carboxylate 3 (49.2 g, 0.21 mol, 1.00 equiv.), spirolactam 4 (39.2 g, 0.23 mol, 1.10 equiv.), DIPEA (54.7 g, 0.42 mol, 2.00 equiv.), and DMAc (147.6 mL, 3 vol). The batch was heated to 90-95 °C, and after 60 h, IPC confirmed -14% (AUC) of ethyl 4-chloro-2-(methylthio)pyrimidine-5-carboxylate remained. The batch was cooled to RT, and precipitate formation was observed. The suspension was diluted with MTBE (100 mL, 2 vol) and water (442 mL, 9 vol) and stirred for 2 h at RT. The product was isolated by vacuum filtration and washed with MTBE (49 mL, 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford compound 5 [41.0 g, 53% yield] as an off-white solid with a purity of >99% AUC. ¾ MR (CDCh): δ 8.76 (d, J = 2.0 Hz, 1H), 6.51-6.29 (br, 1H), 4.33 (q, J = 7.0 Hz, 2H), 3.78 (s, 2H), 3.58 (s, 2H), 2.92 (s, 2H), 2.53 (s, 3H), 1.63-1.37 (m, 12H). LCMS (ESI, m/z = 365.3 [M+H]).
Preparation of Compound 6:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 5 [41.0 g, 0.11 mol, 1.00 equiv.], Boc-anhydride (36.8 g, 0.17 mol, 1.50 equiv.), DMAP (1.37 g, 0.01 mol, 0.10 equiv.), and dichloromethane (287 mL, 7 vol). The batch was stirred for 3 h at RT. IPC confirmed no starting material remained (AUC). The batch was concentrated into a residue under reduced pressure and taken to the next step (a quantitative yield is assumed for this step). An aliquot (200 mg) was purified by column chromatography (heptanes/ethyl acetate 0 to 100%) to afford compound 6. 1H MR (CDCh): δ 8.64 (s, 1H), 4.31 (q, J = 7.0 Hz, 2H), 4.07 (s, 2H), 3.83 (S, 2H), 3.15 (m, 2H), 2.56 (s, 3H), 172 (m, 3H), 1.59 (m, 15H), 1.42 (t, J= 7.0 Hz, 3H). LCMS (ESI, m/z = 465.2 [M+H]).
Preparation of Compound 7:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with compound 6 [residue from a previous step, quantitative yield assumed, 52.2 g, 0.11 mol, 1.00 equiv.], and THF (261 mL, 5 vol). The batch was cooled to 0°C and 1,8-diazabicyclo[5.4.0]un-dec-7-ene (17.1 g, 0.11 mmol, 1.00 equiv.) was added keeping the internal temperature in 0-10°C range. After the addition was complete, the cooling bath was removed and the reaction mixture was allowed to warm up to RT and after 2 h, IPC confirmed no starting material remained. The batch was seeded with the product (1.0 g) and was cooled to 0°C. The slurry was stirred at 0°C for 2 h. The product was isolated by vacuum filtration and washed with cold (0°C) THF (50 mL, 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40°C for 16 h to afford 7 [47 g, quantitative yield] as a light orange solid with a purity of >99% AUC. The color of the product changed into yellow once the batch was exposed to air for an extended period of time (~ 1 day). Material was isolated with substantial amount DBU, according to proton NMR. However, it did not interfere with the next step. 1H MR (CDCh): δ 8.71 (s, 1H), 4.03 (s, 2H), 2.57 (s, 3H), 1.85 (m, 10H), 1.51 (s, 9H). LCMS (ESI, m/z = 419.2 [M+H]).
Preparation of Compound 8:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 7 [40.8 g, 0.10 mol, 1.00 equiv.], triethylamine (31.5 g, 0.31 mol, 3.20 equiv.), and dichloromethane (408 mL, 10 vol). The batch was purged with N2 for 15 min and was cooled to 0°C. Triflic anhydride (44.0 g, 0.16 mol, 1.60 equiv.) was added keeping the
internal temperature in 0-10°C range. The batch was stirred at 0°C and after 3 h, IPC confirmed -7.0% (AUC) of 7 remained. [It was speculated that the product was hydrolyzing back into starting material during the analysis.] Once the reaction was deemed complete, the batch was transferred to a 1 L, separatory funnel and was washed with 50% saturated sodium bicarbonate (200 mL, 5 vol). [It was prepared by mixing saturated sodium bicarbonate (100 mL) with water (100 mL)).] The aqueous layer was separated and was extracted with DCM (2×40 mL, 1 vol). The organic layers were combined and concentrated into a residue under reduced pressure and taken to the next step. LCMS (ESI, m/z = 551.6 [M+H]).
Preparation of Compound 9:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with compound 8 [residue from a previous step, quantitative yield assumed, 53.7 g, 0.10 mol, 1.00 equiv.], and THF (110 mL, 2 vol). The solvent was removed under vacuum distillation and the procedure was repeated two times. The flask was charged with triethylsilane (22.7 g, 0.20 mol, 2.00 equiv.), and DMF (268 mL, 5 vol). The batch was degassed by five cycles of evacuation, followed by backfilling with nitrogen. The flask was charged with tetrakis(triphenylphosphine)palladium(0) (11.3 g, 0.01 mol, 0.1 equiv.). The batch was heated to 45-50°C, and after 14 h, IPC confirmed no starting material remained. The batch was transferred to a 500 mL, separatory funnel while still warm. The reaction was partitioned between water (5 vol) and ethyl acetate (5 vol). The aqueous layer was extracted with ethyl acetate (3 x3 vol). The organic layers were combined and concentrated down to 2 volumes. The precipitate was filtered and washed with ethyl acetate (2x 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40°C for 16 h to afford 9 [27.5 g, 70% yield] as a yellow solid with a purity of -98% AUC. Proton NMR showed some triphenylphosphine oxide present. ¾ NMR (DMSO-i¾):5 9.01 (s, 1H), 7.40 (s, 1H), 4.30 (s, 2H), 2.58 (m, 2H), 2.58 (s, 3H), 1.81 (m, 5H), 1.51 (s, 9H). LCMS (ESI, m/z = 403.4 [M+H]).
Preparation of Compound 10 from the Scheme 2-1 route:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged 9 (12.8 g, 31.8 mmol, 1.00 equiv.) and dichloromethane (64 mL, 5 vol). Trifluoroacetic acid (18.2 g, 159 mmol, 5.00 equiv.) was added over 20 min and the solution was stirred for 2 h at RT. IPC confirmed reaction was complete. The batch was transferred to a 500 mL, separatory funnel and washed with saturated sodium bicarbonate (200 mL). The aqueous layer was extracted with dichlorom ethane (3 x3 vol). The organic layers were combined and concentrated down to 1 volume. The precipitate was filtered and conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford 9 [6.72 g, 70% yield] as an off-white solid with a purity of 99.1% AUC. ¾ NMR (DMSO-dis): δ 8.95 (s, 1H), 8.32 (s, 1H), 7.15 (s, 1H), 3.68 (d, J = 1.0 Hz, 2H), 2.86 (m, 2H), 2.57 (s, 3H), 1.92 (m, 2H), 1.73 (m, 3H), 1.39 (m, 3H). LCMS, ESI, m/z = 303.2 [M+H]).
Preparation of Compound 10 from Scheme 2-2 route:
A 50 mL, three-neck flask equipped with a magnetic stirring bar, thermocouple, N2 inlet was charged 14 (680 mg, 1.62 mmol, 1.00 equiv.) and THF (6.8 mL, 10 vol). A I M solution of potassium tert-butoxide (3.2 mL, 3.24 mmol, 2.00 equiv.) in THF was added over 10 min and the solution was stirred for 2 h at RT. IPC confirmed reaction was complete. The batch was acidified with 4 N hydrogen chloride solution in dioxane (2.4 mL, 9.72 mmol, 6.00 equiv.) and stirred for additional 1 h. The batch was transferred to a 500 mL, separatory funnel and washed with saturated sodium bicarbonate (100 mL). The aqueous layer was extracted with ethyl acetate (3 x20 vol). The organic layers were combined and concentrated down to 3volumes and product precipitated. The precipitate was filtered and conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford 9 [489 mg, quantitative yield] as an off-white solid.
Preparation of Compound 11 :
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 10 (9.00 g, 29.8 mmol, 1.00 equiv.), and acetonitrile (180 mL, 20 vol). A solution of Oxone (45.9 g, 0.15 mol, 5.00 equiv.) in water (180 mL, 20 vol) was added to the batch over 20 min. The batch was stirred for 2 h and IPC confirmed the reaction was complete. The batch was concentrated down to ½ of the original volume and was extracted with dichloromethane DCM (4x 10 vol). The organic layers were combined; polish filtered and concentrated down to -10 vol of DCM. The product was slowly crystallized out by addition of heptanes (-30 vol). The mixture was cooled to 0°C and the product was filtered and dried under vacuum at 40 °C for 16 h to afford 11 [9.45 g, 95% yield] as an off-white solid with a purity of >99% AUC. ¾ NMR (CDCb): 5 9.24 (s, 1H), 7.78 (s, 1H), 7.46 (s, 1H), 3.89 (d, J= 2.0 Hz, 2H), 3.43 (s, 3H), 2.98 (m, 2H), 2.10 (m, 2H), 1.86 (m, 3H), 1.50 (m, 3H). LCMS (ESI, m/z = 335.2 [M+H]).
Preparation of Compound 13:
A 250 mL, single-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet, and reflux condenser was charged with 4-chloro-2-(methylthio)pyrimidine-5-carbaldehyde (2.00 g, 10.6 mmol, 1.00 equiv.), spirolactam 4 (1.96 g, 11.7 mmol, 1.10 equiv.), DIPEA (2.74 g, 21.2 mmol, 2.00 equiv.), and fert-butanol (20 mL, 10 vol). The batch was heated to 80-85 °C, and after 24 h, IPC confirmed no aldehyde 12 remained. The batch was cool to RT and concentrated into a residue, which was loaded on silica gel column. The product was eluted with ethyl acetate/heptanes (0% to 100%). The product containing fractions were pulled out and concentrated to afford 13 [0.98 g, 29% yield] as an off-white solid.
Preparation of Compound 14:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 13 [0.98 g, 3.00 mmol, 1.00 equiv.], Boc-anhydride (4.90 g, 21.5 mmol, 7.00 equiv.), DMAP (36 mg, 0.30 mmol, 0.10 equiv.), and dichloromethane (7 mL, 7 vol). The batch was stirred for 3 h at RT. IPC confirmed no starting material remained. The batch was cool to RT and concentrated into a residue, which was loaded on silica gel column. The product was eluted with ethyl acetate/heptanes (0% to 100%). The product containing fractions were pulled out and concentrated to afford 14 [0.98 g, 29% yield] as an off-white solid.
Preparation of Compound 15:
To a suspension of methyl glycinate (500 g, 3.98 mol, 1 eq) in DCM (10 L) was added
TEA dropwise at rt under nitrogen atmosphere, followed by the addition of cyclohexanone (781 g, 7.96 mol, 2 eq) dropwise over 15 min. To the resulting mixture was added TMSCN (591 g, 5.97 mol, 1.5 eq) dropwise over 1 hour while maintaining the internal reaction temperature below 35
°C. After stirred at rt for 2 hrs, the suspension became a clear solution. The progress of the reaction was monitored by H- MR.
When the methyl glycinate was consumed completely as indicated by H-NMR analysis, the reaction was quenched by water (5 L). The layers were separated. The aqueous layer was extracted with DCM (1 L). The combined organic phase was washed with water (5 L X 2) and
dried over Na2S04 (1.5 Kg). After filtration and concentration, 1.24 Kg of crude 15 was obtained as oil.
The crude 15 was dissolved in IPA (4 L). The solution was treated with HC1/IPA solution (4.4 mol/L, 1.1L) at RT. A large amount of solid was precipitated during the addition. The resulting suspension was stirred for 2 hrs. The solid product was collected by vacuum filtration and rinsed with MTBE (800 mL). 819 g of pure 15 was obtained as a white solid. The yield was 88.4%. ¾- MR (300 MHz, CD3OD) 4.20 (s, 2H), 3.88 (s, 3H), 2.30-2.40 (d, J = 12 Hz, 2H), 1.95-2.02 (d, J = 12 Hz, 2H), 1.55-1.85 (m, 5H), 1.20-1.40 (m, 1H).
Preparation of Compound 16:
To a solution of 15 (10 g, 43 mmol) in MeOH (100 mL) was added methanolic hydrochloride solution (2 .44 mol/L, 35.3 mL, 2 eq) and Pt02 (0.5 g, 5 wt %). The reaction suspension was stirred with hydrogen bubble at 40 °C for 6 hours. H- MR analysis showed consumption of 15. To the reaction mixture was added K2CO3 (15 g, 108 mmol, 2.5 eq) and the mixture was stirred for 3 hrs. The suspension was filtered and the filtrate was concentrated to dryness. The residual oil was diluted with DCM (100 mL) and resulting suspension was stirred for 3 hrs. After filtration, the filtrate was concentrated to provide crude 16 (6.6 g) as an oil. The crude 16 was diluted with EtOAc/hexane (1 : 1, 18 mL) at rt for 2 hrs. After filtration, 16 (4 g) was isolated. The obtained 16 was dissolved in DCM (16.7 mL) and hexane (100 mL) was added dropwise to precipitate the product. After further stirred for 1 h, 2.8 g of the pure 16 was isolated as a white solid. The yield was 39%. HPLC purity was 98.3%; MS (ESI): 169.2 (MH+); 1 H-NMR (300 MHz, D2O) 3.23 (s, 3H), 3.07 (s, 3H), 1.37-1.49 (m, 10H).
Preparation of compound 19:
5-(4-methylpiperazin-l-yl)pyridin-2-amine (803.1 g; 3.0 equivalents based on sulfone 11) was charged to a 22 L flask. The flask was blanketed with N2 and anhydrous THF added (12.4 kg). The resulting black-purple solution was cooled in an ice bath to < 5°C. 1M LiHMDS (4.7 L; 1.2 equivalents based on sulfone 11) was added via an addition funnel in three equal additions to keep the temperature below 10°C. Upon the completion of the addition, the reaction mixture was warmed to 16°C. The sulfone 11 (455.1 g; 1.00 equivalents) was added in five additions. Reaction proceeded until HPLC analysis of an IPC sample indicated less than 3% of sulfone 11 remained.
To quench the reaction, the contents of the 22L flask were transferred to a 100 L flask containing water. After stirring for 30 minutes at <30°C, the crude product was collected by filtration, washed with water and dried to afford 19 (387 g, 99.1% purity, 63.7% yield).
Preparation of compound 20:
5-(4-isopropylpiperazin-l-yl)pyridin-2-amine (1976.2 g; 3.0 equivalents based on sulfone 11) was charged to a 50 L flask. The flask was blanketed with N2 and anhydrous THF added (10.7 kg). The resulting black-purple solution was cooled in an ice bath to < 5°C. 1M LiHMDS (9.6 kg; 3.6 equivalents based on sulfone) was added via an addition funnel at a rate to keep the temperature below 10°C. Upon the completion of the addition, the reaction mixture was warmed to 16°C over 120 minutes by removing the ice bath. The sulfone (1000 g; 1.00 mol) was added in five additions. The reaction proceeded until HPLC analysis of an IPC sample indicated less than 1% of sulfone 11 remained. After completion of the reaction, ammonium chloride was added to the reaction mixture. The mixture stirred at < 32°C for at least 30 minutes and the solids collected by filtration to afford 20 (900 g, 99.1% purity, 64.2% yield).
Alternate Route to Spirolactam via cyclohexanone:
Scheme 2-7
26
In one embodiment the spirolactam is made via the synthetic scheme above. By reducing the nitrile group before addition of the glycinate group the reaction sequence proceeds in higher yield. The chemistry used in Step 1 is described in the literature (J. Org. Chem. 2005, 70,8027-8034), and was performed on a kilogram scale. The chemistry to convert Compound 24 into the
spirolactam was also demonstrated on kilogram scale. The Boc protection of Compound 23, is carried out at -70°C in order to limit formation of the di-Boc protected product. Experimental details of a 200 g pilot run are described below.
Step 1
200 g of cyclohexanone 21 was converted to 22 using Ti(Oi-Pr)4 /TMSCN/NH3. After work-up, 213 g of 22 was obtained. The H- MR was clean. The yield was 84%. The titanium salts were removed by vacuum filtration. In one embodiment, the titanium salts are removed by centrifugation or Celite filtration.
Step 2
190 g of 22 was mixed with LAH (2 eq) in MTBE for 30 minutes at 45°C. After work-up, 148 g of crude 23 was obtained.
Step 3
136 g of the crude 23 from step 2 was converted to 24 with 0.9 eq of B0C2O at -70°C. The reaction was completed and worked up. After concentration, 188 g of 24 was obtained. The yield was 86%. The H-NMR and C-NMR spectra confirmed that the compound was pure.
Step 4
188 g of 24 was subjected to methyl 2-bromoacetate and K2CO3 in acetonitrile to afford 25. 247 g of crude 25 was obtained.
Step 5
247 g of 25 was subjected to TFA in DCE heated to reflux to afford 26. After work-up, 112 g of 6 as TFA salt was obtained. H- MR was clean.
Step 6
26 27
Compound 26 was stirred in EtOH in the presence at room temperature overnight to afford 27. In one embodiment DCM is used as the solvent instead of EtOH.
Example 3. Purge of residual palladium from Step 5 Scheme 2-1:
Since palladium was used in Step 5 of Scheme 2-1, the levels of residual Pd present in the subsequent synthetic steps was determined. Table 2 below and Figure 3 show the palladium levels in the isolated solids.
Table 2
The material after Step 5 was isolated containing 1.47% (14700 ppm) of residual palladium. This data represents the highest level of palladium in the worst case scenario. The workup conditions of the latter steps purged nearly all of the palladium and the final product, 19 bis HC1 salt, contained 14 ppm of Pd, which is below the standard 20 ppm guidline. The Pd levels will likely be even lower once the catal st loading is optimized in Step 5.
19
The process developed in this route was a significant improvement over the one used for the first generation synthesis. Overall, the scheme consists of seven steps with five isolations, all by crystallization. No silica column chromatography is employed in the synthesis, which makes the process highly scalable. The process workup conditions can successfully purge the 1.47% of residual palladium after step 5 of Scheme 2-1.
Nastorazepide, also known as Z-360, is a selective, orally available, 1,5-benzodiazepine-derivative gastrin/cholecystokinin 2 (CCK-2) receptor antagonist with potential antineoplastic activity. Z-360 binds to the gastrin/CCK-2 receptor, thereby preventing receptor activation by gastrin, a peptide hormone frequently associated with the proliferation of gastrointestinal and pancreatic tumor cells.
In January 2018, Zeria is developing nastorazepide calcium (phase II clinical trial), a CCK2 receptor antagonist, for the treatment of pancreatic cancer.
Zeria is developing nastorazepide calcium (Z-360), an oral CCK2 receptor (gastrin receptor) antagonist, for the potential treatment of pancreatic cancer. In September 2005, a phase Ib/IIa trial began in the UK for pancreatic cancer , in February 2008, the trial was completed ; in June 2008, data were presented . In March 2010, the drug was listed as being in phase II preparation in Europe ; in August 2011, this was still the case . In April 2014, a phase II trial began in patients with metastatic pancreatic adenocarcinoma in Japan, Korea and Taiwan. In November 2015, the drug was listed as being in phase II development
Nastorazepide (calcium salt)
CAS No. : 343326-69-2
M.Wt:540.62Formula:C29H36N4O5Ca0.5
Cholecystokinin (CK) is a digestive hormone produced and released in the duodenum, jejunal membrane and is known to have actions such as secretion of secretion, constriction of the gallbladder, stimulation of insulin secretion and the like. C CK is also known to exist in high concentrations in the cerebral cortex, hypothalamus and hippocampus, and it is also known that it has actions such as suppression of food intake, memory enhancement, anxiety action and the like. On the other hand, gastrin is a gastrointestinal hormone produced and released in G cells distributed in the pyloric region of the stomach, and it is known that it has gastric acid secretion action, contraction action of the gastric pyloric part and gallbladder, and the like. These C CK and gastrin have the same 5 amino acids at the C-terminus, and all express the action through the receptor. C CK receptors are classified into peripheral type C CK – A distributed in the ile, gall bladder and intestinal tract and central type C CK – B distributed in the brain. The gastrin receptor and the CKK – B receptor show similar properties in receptor binding experiments and sometimes called C CK 1 B / gastrin receptor due to high homology. These receptors, such as gastrin or a CCK-B receptor antagonist compound, are useful in the treatment of gastric ulcers, duodenal ulcers, gastritis, reflux esophagitis, splenitis, Zollinger-EUison syndrome, cavitary G cell hyperplasia, basal hyperplasia, Choleditis, gallstone stroke, gastrointestinal motility disorder, sensitive bowel syndrome, certain tumors, eating disorders, anxiety, panic disorder, depression, schizophrenia, Parkinson’s disease, late onset dyskinesia, It is expected to be useful for treatment and prevention of La Tourette’s syndrome, addiction due to drug ingestion, and withdrawal symptoms. It is also expected that the induction of analgesia or the enhancement of induction of analgesia by opioid drugs is expected (Journal of Pharmacology, Vol. 106, 171-180 (1995), Drugs of the Future, Vol. 18, 919-931 (1993), American Journal of Physiology, Vol.
As a gastrin receptor antagonist already, prolumide is known as a therapeutic agent for gastric ulcer and gastritis. However, proglumide has considerably low affinity for gastrin or CKK-B receptor and its therapeutic effect is weak. In addition, L – 3 6 4, 7 1 8 (Dibazepide, Japanese Unexamined Patent Publication No. 616366), L -3 6 5, 2 6 0 (Japanese Patent Laid-Open No. 6 3- 9), and the like, have been reported to exhibit either CKK-A receptor antagonism or CKK-B receptor antagonism. Furthermore, it is disclosed that a compound having a strong C 4 C – – B receptor antagonistic effect suppresses gastric acid secretion by pentagastrin stimulation (International Patent Publication WO 94/438, International Patent Publication WO 95/18110) , It is not always satisfactory and clinically applicable gastrin or CKK-B receptor antagonist has not yet been provided.
Compounds capable of strongly binding to gastrin or cholecystokinin receptors are expected for the prevention and treatment of diseases involving their respective receptors in the digestive tract and the central nervous system.
Compound A ((R) – (-) – 3- [3- (1-tert-butylcarbonylmethyl-2-oxo-5-cyclohexyl- 1,3,4,5-tetrahydro- 2H- 1,5-benzodiazepine -3-yl) ureido] benzoate) has the following structural formula and can be produced by the method described in Patent Document 1.
[Chemical formula 1]
Example 1
Compound A 20.0 g of amorphous substance was suspended in 253 mL of methanol. After dissolving by heating, it was cooled and the precipitated crystals were collected by filtration and washed with methanol. The obtained wet crystals were dried under reduced pressure.
1 H-NMR (DMSO-d 6 ) δ: 1.18 (18H, s), 1.10-2.03 (20H, m), 3.17 (12H, d), 3.19-3.29 (4H, m), 3.37-3.44 (2H, (2H, m), 7.07-7.12 (2H, m), 4.07-4.16 (4H, br)
IR (KBr) cm -1 : 2935 (2H, m), 7.15 (2H, t), 7.22-7.29 (4H, m), 7.50-7.56 (4H, m), 7.88 , 2361, 1648, 1553, 1497, 1388, 1219, 776
The powder X-ray diffraction spectrum of the obtained crystal is shown in FIG. 2. From NMR, IR and FIG. 2, the obtained crystals were Compound AI type crystals.
Example 5
Compound A 50.0 g of amorphous material was suspended in 380 mL of isopropanol (IPA). After dissolving by heating, it was cooled and precipitated. Precipitated crystals were collected by filtration and washed with IPA to obtain wet crystals. This was dried under reduced pressure. The powder X-ray diffraction spectrum of the obtained crystal is shown in FIG. 1 H-NMR (DMSO-d 6 ) [delta]: 1.04 (24H, d), 1.18 (18H, s), 1.10-2.03 (20H, m), 3.16-3.28 (4H, m), 3.37-3.45 (2H, (2H, m), 7.07-7.12 (2H, m), 3.72-3.83 (4H, m), 4.33-4.43 (8H, m), 5.13 (2H, d), 6.71
IR (KBr) cm -1 : 2933 (2H, m), 7.15 (2H, t), 7.21-7.30 (4H, m), 7.48-7.54 (4H, m), 7.84 , 2361, 1653, 1553, 1498, 1394, 1219, 769
From NMR, IR and FIG. 4, the obtained crystals were Compound AIII type crystals.
Process for producing a calcium salt of a 1,5-benzodiazepine compound – nastorazepide calcium – a cholecystokinin CCK2 receptor antagonist. Useful for the treatment of gastritis, reflux esophagitis, Zollinger-Ellison syndrome.
Example 1
(1) (R) – (-) – 2-Oxo-3-tert-butoxycarbonylamino-5-cyclohexyl-1,3,4,5-tetrahydro-2H-1,5-benzodiazepine (compound 2)), 139.3 g of 1-chloropinacolone and 8.3 g of tetrabutylammonium bromide in 1432 ml of toluene was added dropwise 461 g of 30% sodium hydroxide aqueous solution at 10 ° C. or lower. After stirring for 1 hour, the aqueous layer was removed. To the toluene layer, 620 ml of water was added and the liquid was separated, and the toluene layer was used for the next step.
(2) 628.9 g of hydrochloric acid was added dropwise to the toluene layer obtained in the previous step at 30 ° C. or lower. After stirring for 30 minutes, liquid separation was carried out, and the aqueous layer was separated. It was neutralized with 908.5 g of 30% sodium hydroxide aqueous solution and extracted with 1432 ml of toluene. The toluene layer was separated with 620 g of a 20% sodium chloride aqueous solution, and toluene was distilled off under reduced pressure. (R) – (-) – 1 -tert-butylcarbonylmethyl-2-oxo-3-amino-5- cyclohexyl-1,3,4,5-tetrahydro-2H-1,5-benzodiazepine (Compound (6) ) Was obtained.
(3) The (R) – (-) – 1-tert-butylcarbonylmethyl-2-oxo-3-amino-5-cyclohexyl-1,3,4,5-tetrahydro-2H-1 , 5-benzodiazepine (Compound (6)), 221.8 g of 3-phenyloxycarbonylaminobenzoic acid, 174.5 g of triethylamine and 77.7 g of water were added and the mixture was stirred at 45 to 50 ° C. for 2 hours. To the reaction solution were added 1375 ml of ethanol and 930 ml of water, and 62.9 g of hydrochloric acid was added dropwise at 30 ° C. or lower. The precipitated crystals were centrifuged.
The obtained crystals were heated to dissolve in 4714 ml of ethanol at 60 ° C., and 2790 ml of water was added dropwise to precipitate crystals. The precipitated crystals were separated by centrifugation and dried under reduced pressure to give (R) – (-) – 3- [3- (1-tert-butylcarbonylmethyl-2-oxo-5-cyclohexyl- 5-tetrahydro-2H-1,5-benzodiazepin-3-yl) ureido] benzoic acid (Compound (5)) 0.5 ethanolate monohydrate 430.2 g.
(4) (R) – (-) – 3- [3- (1-tert-Butylcarbonylmethyl-2-oxo-5-cyclohexyl-1,3,4,5-tetrahydro-2H- 1,5-benzodiazepine -3-yl) ureido] benzoic acid (Compound (5)) 0.5 Ethanol solvate monohydrate 430.3 g was suspended in 1645 ml of isopropyl alcohol (IPA), sodium hydroxide 31.6 g / A solution of 934 ml of water was added dropwise to dissolve (a).
112.7 g of calcium chloride dihydrate was dissolved in 3012 ml of water. Here, the solution of (a) was added dropwise at 10 ° C. or lower. After dropping, the temperature was raised to 50 ° C., after stirring for 2 hours, it was cooled to 10 ° C. or lower. The precipitated powder was centrifuged and washed with a mixed solution of IPA 658 ml / water 2065 ml, followed by 4303 ml of water and dried under reduced pressure to give (R) – (-) – 3- [3- (1-tert- Oxo-5-cyclohexyl-1,3,4,5-tetrahydro-2H-1,5-benzodiazepin-3-yl) ureido] benzoate (compound (1)). The powder X-ray diffraction spectrum was measured (as 7% water content), and the obtained compound (1) was amorphous.
Example 2 In
step (4) of Example 1, investigation was carried out by changing the amount of the solvent and sodium hydroxide.
First, when the IPA / water ratio is 1 / 2.5 to 1/10, preferably 1 / 2.75 to 1/8, more preferably 1 / 2.75 to 1/5, the compound (1 ) Amorphous can be stably obtained.
Next, when the amount of sodium hydroxide is 1.0 to 1.10 mol with respect to the compound (1) and the amount of calcium chloride is 0.5 to 1.5 mol with respect to the compound (1), the amount of the compound 1) can be obtained in high yield.
Further, it was found that impurities are not produced when the reaction temperature of the compound (1) and sodium hydroxide in the step (4) is 20 ° C. or less, more preferably 10 ° C. or less, further preferably 0 to 10 ° C.
METHOD FOR MANUFACTURING 1, 5-BENZODIAZEPINE DERIVATIVE
2012-01-12
1: Kato H, Seto K, Kobayashi N, Yoshinaga K, Meyer T, Takei M. CCK-2/gastrin receptor signaling pathway is significant for gemcitabine-induced gene expression of VEGF in pancreatic carcinoma cells. Life Sci. 2011 Oct 24;89(17-18):603-8. doi: 10.1016/j.lfs.2011.07.019. Epub 2011 Aug 3. PubMed PMID: 21839751.
////////////NASTORAZEPIDE, phase II, treatment of pancreatic cancer,
Phase II Breast cancer; Prostate cancer; Solid tumours
31 Jan 2019 Innocrin Pharmaceutical completes a phase II trial in Prostate Cancer (Second-line therapy or greater, Hormone refractory) in the US (NCT02445976)
31 Jan 2019 Innocrin Pharmaceutical completes a phase II trial for Prostate Cancer (Hormone refractory) in the US, UK, Switzerland and Greece (NCT02012920)
31 Jan 2019 Innocrin Pharmaceuticals completes the phase I/II CLARITY-01 trial for Breast cancer (Late stage disease) in USA (NCT02580448)
CYP-17 useful for treating fungal infections, prostate cancer, and polycystic ovary syndrome, assigned to Viamet Pharmaceuticals Inc , naming Hoekstra and Rafferty. Innocrin Pharmaceuticals , a spin-out of Viamet is developing oral seviteronel, the lead dual selective inhibitors of the 17,20-lyase activity of P450c17 (CYP17) and androgen receptor antagonist, which also includes VT-478 and VT-489, developed using the company’s Metallophile technology, for treating castration-resistant prostate cancer (CRPC) in men, breast cancer and androgen (AR) related cancers.
The present invention relates to a process for preparing compound 1 that is useful as an anticancer agent. In particular, the invention seeks to provide a new methodology for preparing compound 1 and substituted derivatives thereof.
Living organisms have developed tightly regulated processes that specifically import metals, transport them to intracellular storage sites and ultimately transport them to sites of use. One of the most important functions of metals such as zinc and iron in biological systems is to enable the activity of metalloenzymes. Metalloenzymes are enzymes that incorporate metal ions into the enzyme active site and utilize the metal as a part of the catalytic process. More than one-third of all characterized enzymes are metalloenzymes.
The function of metalloenzymes is highly dependent on the presence of the metal ion in the active site of the enzyme. It is well recognized that agents which bind to and inactivate the active site metal ion dramatically decrease the activity of the enzyme. Nature employs this same strategy to decrease the activity of certain metalloenzymes during periods in which the enzymatic activity is undesirable. For example, the protein TIMP (tissue inhibitor of metalloproteases) binds to the zinc ion in the active site of various matrix metalloprotease enzymes and thereby arrests the enzymatic activity. The pharmaceutical industry has used the same strategy in the design of therapeutic agents. For example, the azole antifungal agents fluconazole and voriconazole contain a l-( 1,2, 4-triazole) group that binds to the heme iron present in the active site of the target enzyme lanosterol demethylase and thereby inactivates the enzyme.
In the design of clinically safe and effective metalloenzyme inhibitors, use of the most appropriate metal-binding group for the particular target and clinical indication is critical. If a weakly binding metal-binding group is utilized, potency may be suboptimal. On the other hand, if a very tightly binding metal-binding group is utilized, selectivity for the target enzyme versus related metalloenzymes may be suboptimal. The lack of optimal selectivity can be a cause for clinical toxicity due to unintended inhibition of these off-target metalloenzymes.
One example of such clinical toxicity is the unintended inhibition of human drug metabolizing enzymes such as CYP2C9, CYP2C19 and CYP3A4 by the currently-available azole antifungal agents such as fluconazole and voriconazole. It is believed that this off-target inhibition is caused primarily by the indiscriminate binding of the currently utilized l-(l,2,4-triazole) to iron in the active site of CYP2C9, CYP2C19 and CYP3A4. Another example of this is the joint pain that has been observed in many clinical trials of matrix metalloproteinase inhibitors. This toxicity is considered to be related to inhibition of off-target metalloenzymes due to indiscriminate binding of the hydroxamic acid group to zinc in the off-target active sites.
Therefore, the search for metal-binding groups that can achieve a better balance of potency and selectivity remains an important goal and would be significant in the realization of therapeutic agents and methods to address currently unmet needs in treating and preventing diseases, disorders and symptoms thereof. Similarly, methods of synthesizing such therapeutic agents on the laboratory and, ultimately, commercial scale is needed. Addition of metal-based nucleophiles (Zn, Zr, Ce, Ti, Mg, Mn, Li) to azole-methyl substituted ketones have been effected in the synthesis of voriconazole (M. Butters, Org. Process Res. Dev. 2001, 5, 28-36). The nucleophile in these examples was an ethyl-pyrimidine substrate. Similarly, optically active azole-methyl epoxide has been prepared as precursor electrophile toward the synthesis of ravuconazole (A. Tsuruoka, Chem. Pharm. Bull. 1998, 46, 623-630). Despite this, the development of methodology with improved efficiency and selectivity is desirable
Preparation of Compound 4:
de
Acetone (850 L), 2,3-dihydroxynaphthalene (85.00 kg, 530.7 moles), and potassium carbonate (219.3 kg, 1,586.7 moles) were charged to a clean, fixed reactor with stirring and with the temperature maintained at 20 – 35 °C. Dimethyl sulfate (200.6 kg, 2131.09) was added to the stirred reaction at a rate that maintains the internal temperature of the exothermic reaction below 60 °C. This addition typically requires about 3 hours. At the end of the dimethyl sulfate addition, the reaction is continued to allow to stir while maintaining the internal temperature at 50 – 60 °C. After about 3 hours, the reaction was analyzed by HPLC. The reaction was concentrated by atmospheric pressure distillation of acetone. The distillation was continued until 340 – 425 L of distillate was collected. This represents 40 – 50 % of the initial charge of acetone. At the end of the distillation, the reaction mass is present as a thick suspension. While maintaining the internal temperature below 60 °C, the reactor contents were slowly diluted with water (850 L). When the addition is complete, the reaction was cooled to an internal temperature of 25 – 35 °C and stirring was continued for 1 – 2 hours after the designated internal temperature was reached. Compound 2 was isolated by filtration and the cake was washed with water (at least 3 X 85 L). Compound 2 was dried at 40 – 45 °C and full vacuum until the water content by Karl Fisher titration is found to be NMT 2.0 %. Typically, greater than 90 kg of dry product is obtained with an assay of >99.5% AUC by HPLC.
Dichloromethane (with a water content by Karl Fisher Titration of NMT 0.50%) (928 L) and 2,3-dimethoxynaphthalene (2, 116.00 kg, 616.3 moles) were charged to a clean, fixed reactor with stirring and with the temperature maintained at 20 – 35 °C. The reactor contents were cooled to an internal temperature of -5 to 0 °C. Aluminum chloride (164.72 kg, 1235.3 moles, 2.00 molar equivalents) was carefully added in portions to the reaction, while maintaining the internal temperature at -5 to +5 °C. This addition typically requires 5 – 6 hours. At the end of the addition, the reactor contents were cooled to an internal temperature of -15 to -5 °C. Isobutyryl chloride (102.08 kg, 958.05 moles, 1.55 molar equivalents) was slowly added to the reaction while maintaining the internal temperature at -15 to -5 °C. The addition typically requires about 3 hours. At the end of the isobutyryl chloride addition, the reaction was warmed to an internal temperature of 20 – 35 °C. When the temperature was reached, these conditions were maintained for 2 – 3 hours until the IPC indicated a level of residual starting material of NMT 2.0 % AUC by HPLC. The reactor contents were then cooled to 0 – 5 °C. The reaction was quenched by adding the reaction to a precooled (0 – 5 °C) 3M aqueous solution of hydrochloric hcid (Water, 754 L: cone. HC1, 406 L). The mixture was vigorously stirred for 15 – 20 minutes then the layers were allowed to settle. The lower, dichloromethane, product-containing layer was washed sequentially with 10 % aqueous sodium bicarbonate (1044 L), water (1160 L), then 10 % aqueous sodium chloride (1044 L). The reaction was concentrated by distillation under full vacuum and at an internal temperature of NMT 40 °C. The reaction concentrate was cooled to 20 – 35 °C and diluted with hexanes (812 L). The resultant slurry was warmed to 45 – 50 °C and these conditions were maintained for 1 – 2 hours. The reactor contents were cooled to 20 – 35 °C for 1 – 2 hours. Compound 3 was isolated by filtration. The cake was washed with fresh hexanes (232 L) twice, the filter was cooled, and the cake was washed an additional two times with hexanes. Compound 3 was dried under full vacuum at a jacket temperature of 45 °C. Typically, about 95 kg of dry product was isolated with a product purity of >90% by HPLC.
Acetic acid (212.5 L L) and l-(6,7-dimethoxynaphthalene-2-yl)-2-methylpropane-l- one (42.5 kg, 164.5 moles) were charged to a clean, fixed reactor with stirring and with the temperature maintained at 25 – 45 °C. Concentrated hydrochloric acid (425.0 L) was added carefully to the stirring reactor contents while maintaining reactor contents at an internal temperature of 25 – 45 °C. When the addition was complete, the internal temperature of the reaction was raised to 100 – 105 °C. Note that the reaction is a heterogeneous mixture. The reaction was stirred under these conditions for 6 – 8 hours. The reaction was cooled to 85 – 90 °C to which was carefully added a fresh portion of hydrochloric acid (127.5 L). The reaction was warmed to 100 – 105 °C and stirred for another 6 – 8 hours. The reaction was cooled to 85 – 90 °C. The reaction was cooled further to 70 – 80 °C. Water (212.5 L) was added to the well stirred reaction and the reactor contents were cooled to an internal temperature of 35 – 45 °C and stirred for 3 – 4 hours. Compound 4 was collected by filtration. The wet cake was washed with water (212.5 L). The wet cake was added to a clean reactor with a 5% aqueous sodium bicarbonate solution and stirred at an internal temperature of 35 – 45 °C for 1 – 2 hours.
Compound 4 was collected by filtration and washed with water (212.5 L). Compound 4 was dried under full vacuum and a temperature of < 50 °C until the water content of the dried material was found to be NMT 5.0% by Karl Fisher Titration. The yield is typically >31 kg with a purity >99.5 %.
Preparation of Compound 5:
The following difluoromethylation conditions listed in Table 1 were investigated:
Preparation 1:
The reaction flask was dried under an argon flow at 120 °C. (lS,2R)-l-Phenyl-2-(l- pyrrolidinyl)propan-l-ol (ligand 45) (196.6 g, 0.96 mol, 2.2 eq.) was added into the flask and then toluene (195 mL) was added. The solution was cooled to <12 °C. A solution of diethyl zinc (716.4 g, 0.87 mol, 15 wt%, 2 eq.) in toluene was added through a septum over 30 min at 0-10 °C. Further, a solution of ((Trimethylsilyl)ethynyl)-magnesium bromide in THF (1.81 kg; 0.87 mol, 9.7 wt%, 2 eq.) was added over 30 min at 0-10 °C. Finally, trifluoroethanol (87.0 g; 0.87 mol; 2 eq.) was added over 10 min at 0-10 °C. The reaction solution was stirred at 10-12 °C for 3 h. Compound 5 (143.4 g; 0.434 mol; 1 eq.) was added (as a solid) at room
temperature. The reaction mixture was stirred at room temperature for 1 h and at 55 °C for 17 h. The reaction solution was cooled to room temperature and dosed with aqueous HC1 (3600 mL; 7.5 wt%) within 20 min. The temperature of the mixture was kept below 25 °C. Toluene (1250 mL) was added and the mixture was stirred at room temperature for 5 min. The aqueous phase was separated and stored for the recycling of ligand 45. The organic phases were washed with water (638 mL) and concentrated via distillation under reduced pressure (50 mbar). The residue (approx. 184 g) was treated with heptane (200 mL), which was removed
via distillation. The residue was dissolved in heptane (2050 mL) at 50 °C. The mixture was cooled to room temperature and subsequently to -8 °C within 2 hours. The obtained suspension was stirred at -8 °C for 1 h. Crystallized compound 5 (20.0 g; 14%) was isolated via filtration, washed twice with cold (0 °C) heptane (2×20 mL) and dried under vacuum at 50 °C for 12 hours. The combined heptane phases were concentrated under reduced pressure to obtain a 48 wt% solution of compound 18b in heptane (yield: 83.0%). The solution was directly used for the next step.
(7S,2R)-l-Phenyl-2-(l-pyrrolidinyl)propan-l-ol (ligand 45) (13.0 kg, 63.3 mol, 2.2 eq.) was charged into the reactor and toluene (60 L) was added. The solution was cooled to < 12 °C. A solution of diethyl zinc (35.6 kg, 57.3 mol, 20 wt%, 2 eq.) in toluene was added via mass flow controller at 8-16 °C. Further, a solution of ((trimethylsilyl)ethynyl)-magnesium bromide in THF (11.5 kg; 57.3 mol, 9.7 wt%, 2 eq.) was added at 8-16 °C. Finally, trifluoroethanol (5.7 kg; 57.3 mol; 2 eq.) was added over 10 min at 8-16 °C.The reaction solution was stirred at 22-25 °C for 3 h. A solution of compound 5 (9.5 kg; 28.7 mol; 1 eq.) in toluene (20 L) was added at room temperature. The reaction mixture was stirred at 25 °C for 1 h and at 55 °C for 17 h. The reaction solution was cooled to room temperature and dosed in aqueous HC1 (225L; 7.5 wt%) within 20 min. The temperature of the mixture should be kept below 25 °C. Toluene (80 L) was added and the mixture was stirred at room temperature for 5 min. The organic phases was washed with water (50 L) and concentrated via distillation under reduced pressure (50 mbar). The residue was treated with heptane (100 L), which was removed via distillation. The residue was dissolved in heptane (100 L) at 50°C, which was removed via distillation. The residue was dissolved in heptane (25 L). Heptane (110 L) was added, the mixture was cooled to room temperature and subsequently to 0-5 °C and seeded with compound 5 (0.15 kg). The obtained suspension was cooled to -8 °C within 1 h and stirred at this temperature for 2 h. Crystallized compound 5 was removed via filtration. The filtrate was concentrated under reduced pressure to obtain a 48 wt% solution of compound 18b in heptane (calculated 8.8 kg, 71.6%). This solution was directly used for the next step.
Recovery of the chiral ligand ( lS,2R)-l-Phenvl-2-
-l-ol from the
Preparation 1:
The above acidic aqueous phase was diluted with toluene (1000 mL) and the mixture was treated with sodium hydroxide (50 wt% solution) to adjust the pH to 12. The mixture was warmed to 50 °C and sodium chloride (100 g) was added. The aqueous phase was separated and washed with toluene (1000 mL). The combined organic phases were washed with water (200 mL). The combined toluene phases were treated with water (1000 mL) and the pH was adjusted to 2 by the addition of a cone. HC1 solution. The aqueous phase was separated and the mixture was treated with sodium hydroxide (50 wt% solution) at 5 °C to adjust the pH to 12. After seeding, the suspension was stirred at 5 °C for 30 min. The solids were isolated, washed with cold (0 °C) water (4×100 mL) and dried under vacuum at 30 °C for 24 hours. Ligand 45 (178.9g; 91%) was obtained as slightly yellow crystalline solid.
HPLC (purity): 99%.
Preparation 2:
The acidic aqueous phase containing ligand 45 (500 L) was diluted with toluene (125 L) and treated with“Kieselgur” (20 L). The mixture was treated with sodium hydroxide (40 L; 50 wt% solution) to adjust the pH to 12 whereas the temperature was kept <55 °C. The suspension was stirred for 15-20 min and filtered to remove all solids. Toluene (80 L) was added and the aqueous phase was separated. The organic phase was treated with water (150 mL) and the pH was adjusted to 1.5-2 by the addition of an aqueous HC1 solution (10 L; 32 wt%). The aqueous phase was separated, toluene (150 L) was added, and the mixture was treated with sodium hydroxide (5 L; 50 wt% solution) at 5 °C to adjust the pH to 12-12.5. The organic phase was separated, washed with water (30 L), and concentrated under reduced
pressure at 50 °C. Approx. 100L of distillate was removed. A sample of the solution of ligand 45 in toluene was analyzed:
The NMR results indicated a 21.6 wt% solution of ligand 45 in toluene which corresponds to a calculated amount of 118.4 kg (83.6%) of ligand 45.
Preparation of Compound 18a
Preparation 1:
A solution of tertiary alcohol 18b (320 g; 48 wt%; 0.36 mol; 1 eq.) in heptane was dissolved in methanol (800 mL). Potassium carbonate (219 g; 1.58 mol; 4.4 eq.) was added (temperature was kept < 30 °C) and the suspension was stirred at room temperature for 3 h. Water (1250 mL) was added and the mixture was treated with a cone. HC1 solution (approx. 130 mL) to adjust the pH to 7.8. The reaction mixture was extracted twice with methyl- /-butyl ether (MTBE; 2×465 mL). The combined MTBE phases were washed with water (155 mL). Water (190 mL) was added to the MTBE phase and the organic solvent was distilled off under reduced pressure (50 mbar). The obtained emulsion of compound 18a (yield: 99%) was directly used for the next step.
The solution of tertiary alcohol 18b (48 wt%; 57.5 mol; 1 eq.) in heptane was dissolved in methanol (128 L). Potassium carbonate (35.0 kg; 253 mol; 4.4 eq.) was added (temperature was kept < 30 °C) and the suspension was stirred at 20-30 °C for 3 h. Water (200 L) was added and the mixture was treated with an aqueous HC1 solution (approx. 25 L; 32 wt%) to adjust the pH to 7.5 – 7.8. The reaction mixture was extracted twice with MTBE
(2×66.6 L). The combined MTBE phases were washed with water (25 L). Water (30 L) was added to the MTBE phase and the organic solvent was distilled off under reduced pressure (<80 mbar; 55°C). The residue was dissolved in tert-butanol (25 L). The resulting 18a was cooled to <30°C and used directly in the next step.
Benzyl bromide (39.4 g; 0.23 mol; 1 eq.) was dissolved in water (177 mL) and t-BuOH (200 mL). Diisopropylethylamine (DIPEA; 59.4 g; 0.46 mol; 2 eq.) and sodium azide (15.0 g; 0.23 mol; 1 eq.) were added. The suspension was stirred for 5 min at room temperature. A suspension of compound 18a (82 g; 0.23 mol; 1 eq.) in water (123 mL) was treated with t-BuOH (100 mL) and copper (I) iodide (8.8 g; 46 mmol; 0.2 eq.) was added and the temperature was kept below 30 °C. The yellow-brown suspension was stirred for 5 h at room temperature. Zinc powder (5.0 g; 76 mmol) and ammonium chloride (7.4 g; 0.14 mol) were added and the reaction mixture was stirred at room temperature for 3 hours. The mixture was diluted with MTBE (800 mL), water (280 mL), and an aqueous ammonia solution (120 g; 25 wt%). Solids were removed by filtration and additional MTBE (200 mL) and brine (200 mL) were added. The aqueous phase was separated and extracted with MTBE (400 mL). The combined organic phases were treated with water (150 mL) and MTBE was distilled off under reduced pressure (100 mbar). The obtained suspension of compound 31 (113 g; 50 wt%) in water (approx. 113 mL) was directly used for the next step.
Benzyl bromide (11.0 kg g; 64.4 mol; 1,12 eq.) was dissolved in water (40 L) and t-BuOH (60 L). DIPEA (16.4 kg; 126.5 mol; 2,2 eq.) and sodium azide (4.12 kg; 63.3 mol; 1 eq.) were added. The suspension was stirred 5 min at room temperature. A mixture of compound 18a (20.5 kg; 57.5 mol; 1 eq.) in ieri-butanol (see previous step) was added together with water (5 L) and copper (I) iodide (2.2 kg; 11.5 mol; 0.2 eq.) at a temperature < 30 °C. The yellow-brown suspension was stirred for 5 h at room temperature. Zinc powder (1.25 kg; 19 mol, 0.33 eq.) and an aqueous solution of ammonium chloride (2.14 kg; 20 wt%; 40 mol; 0.7 eq.) were added and the reaction mixture was stirred at 20-30 °C for 2 hours. The reaction mixture was concentrated under vacuum (<200 mbar, 55 °C). The residue was diluted with MTBE (200 L), water (30 L), and an aqueous ammonia solution (30 kg; 25 wt%). Solids were removed by filtration over a pad of“Kieselgur NF” (2 kg). Brine (50 L) was added for a better phase separation. The aqueous phase was separated and washed with MTBE (200 L). The combined organic phases were washed with an aqueous HC1 solution (1 N, 52 L) and water (50 L). MTBE was distilled off under reduced pressure (<400 mbar, 55°C; distillate min. 230L). The oily residue was dissolved in ethanol (150 L), which was distilled off under reduced pressure (<300 mbar; 55°C; distillate min. 150-155L) and the residue was dissolved in additional ethanol (60 L). To the resulting solution of compound 31 was added water (24 L) and the mixture was warmed to 50-55 °C. The mixture was cooled to 30 °C and crystallization started. The suspension was stirred at 30 °C for 1 h, cooled to <0 °C within 2 hours, and stirred at -5-0 °C for an additional 2 hours. The solids were isolated and washed with ethanol/water (1/1; v/v) (2 x 12 L). The wet product was dissolved in ethanol (115L) at 60 °C and water (24 L) was added. The mixture was cooled to 40 °C and the crystallization started. The suspension was stirred at 30 °C for 1 h, cooled to <0 °C within 2 hours, and stirred at -5-0 °C for additional 2 hours. The solids were isolated and washed (without stirring) with ethanol/water (1/1; v/v) (3 x 8 L). Pure, wet compound 31 was isolated as a white solid, which was used for the next step without drying. 14.0 kg of wet 31 were obtained with a 31 content of 81.6 wt%. Based on the determined content, the calculated amount of pure 31 was 11.4 kg with a yield of 41% over two steps (from 18b).
Preparation 3: Synthesis of compound 31 directly from compound 18b
Benzyl bromide (1.64 g, 9.59 mmol, 1.12 eq) was dissolved in water (2.4 mL) and
MeOH (2.4 mL). K2CO3 (2.38 g, 17.2 mmol, 2.00 eq), sodium ascorbate (0.34 g, 1.72 mmol, 0.20 eq) and finally sodium azide (0.62 g, 9.40 mmol, 1.10 eq.) were added. The suspension was stirred for 5 min at room temperature. A suspension of 18b (3.08 g; 8.64 mmol, 1.00 eq) in water (2.5 mL) and MeOH (2.5 mL) and the resulting mixture was stirred for 10 min.
CuS04 (0.21 g, 1.30 mmol, 0.15 eq) were added (slightly exothermic reaction). The reaction mixture was stirred for 19 h and the conversion was determined by HPLC (conv. 100%, purity of compound 31 by HPLC: 83 area%). To the yellow-green suspension was added zinc powder (0.24 g, 4.13 mmol, 0.43 eq) and ammonium chloride (0.34 g, 6.36 mmol, 0.74 eq) were added and the reaction mixture was stirred at room temperature for 2 hours. The reaction mixture was concentrated under reduced pressure (150 mbar, 50 °C). The mixture was diluted with MTBE (40 mL), water (15 mL), and an aqueous ammonia solution (6.5 mL). Solids were removed by filtration and brine (5.5 mL) was added. The aqueous phase was separated and extracted with MTBE (20 mL). The combined organic phases were treated with water (10 mL) and the pH was adjusted to a pH of 1 by addition of cone. HC1. After phase separation, the organic layer was washed with water (10 mL). MTBE was distilled off under reduced pressure (100 mbar, 50°C) to give the crude compound 31 as an oil. Water (2.5 mL) and EtOH (30 mL) were added and the mixture was warmed to 50 °C. After cooling to 30 °C, the mixture was seeded with compound 31 and compound 31 started to precipitate. The mixture was kept for 1 h at 30 °C, then cooled to 0 °C over 2 h and kept at 0 °C for 2 h. The resulting product, 31, was collected by filtration and the filter cake was washed with small portions of EtOH/water (1:1). After drying, the product (2.97 g) was obtained as a pale yellow, crystalline solid with an HPLC purity of 79 area% and a NMR content of ca. 70 wt%.
Recrystallization of
31
Preparation 1:
To a suspension of compound 31 (96 g; 0.196 mol; 50 wt%) in water (96 mL) was added ethanol (480 mL) and the mixture was warmed to 50 °C. The mixture was cooled to 30 °C and crystallization started. The suspension was stirred at 30 °C for 1 h, cooled to 0 °C within 2 hours and stirred at 0 °C for additional 2 hours. The solids were isolated and washed with ethanol/water (1/1; v/v) (3 x 40 mL). The wet product was dissolved in ethanol (280 mL) at 60 °C and water (56 mL) was added. The mixture was cooled to 40 °C and crystallization started. The suspension was stirred at 30 °C for 1 h, cooled to 0 °C within 2 hours, and stirred at 0 °C for an additional 2 hours. The solids were isolated and washed with ethanol/water (1/1; v/v) (3 x 28 mL). Pure, wet compound 31 (46.8 g on dried basis; 49 % over 2 steps) was isolated as a white solid, which was used for the next step without drying.
14 kg of ethanol-wet 31 (content 81.6 wt%, calculated 11.4 kg, 23.7 mol) were suspended in ethanol (46 L) and the mixture was warmed to 50-55 °C, forming a homogenous solution at this temperature. Water (9 L) was added at 50-55 °C and the mixture was cooled to 40-45 °C. After the crystallization had started, the suspension was stirred at 40-45 °C for 1 h, cooled to 0 °C within 2 hours, and stirred at 0 °C for additional 2 hours. The solids were isolated and washed with ethanol/water (1/1; v/v) (3 x 8 L). Pure, wet compound 31 (14.5 kg) was isolated as a white solid, which was used for the next step without drying.
Preparation of Azidomethyl Pivalate Protected Triazole (6) from Compound 18a
1
Azidomethyl pivalate (1.42 g, 9.00 mmol, 1.05 eq) was suspended in water (6.0 mL) and t-BuOH (7.2 mL) and the suspension was stirred for 5 min. Compound 18a (theor. 3.08 g, 8.64 mmol, 1.00 eq), sodium ascorbate (0.48 g, 2.4 mmol, 0.30 eq), and CuS04 (0.08 g, 0.40 mmol, 0.05 eq.) were added. The reaction mixture was stirred for 19 h and conversion was determined by HPLC (conv. 98%, purity of the product by HPLC: 81 area%). To the green suspension was added MTBE (20 mL), water (10 mL), and an aqueous ammonia solution (2 g). A biphasic turbid mixture was formed. To improve phase separation, additional MTBE (20 mL) and water (10 mL) were added. The aqueous phase was separated and extracted with MTBE (20 mL). The combined organic phases were concentrated under reduced pressure (100 mbar, 50 °C) to give the crude product as a brown oil that solidified upon standing. HPLC purity: ca. 65 area%; NMR content of ca. 73 wt%.
Preparation of Azidomethyl Pivalate Protected Triazole (6) from 18b
In a reaction flask, sodium ascorbate (277 mg, 1.4 mmol, 1.20 eq) and CuS04 (37 mg, 0.23 mmol, 0.20 eq.) were suspended in MeOH (11 mL). Azidomethyl pivalate (183 mg, 1.16 mmol, 1.00 eq) and 18b (183 mg, 1.16 mmol, 1.00 eq) were added and the mixture was warmed to 60 °C. The reaction mixture was stirred for 19 h and worked up. To the green suspension was added an aq NH4Cl solution (2 mL) and zinc powder, and the mixture was stirred for 2 h. MTBE (2 mL) was added and the aqueous phase was separated and extracted with MTBE (2 mL). The combined organic phases were concentrated under reduced pressure (100 mbar, 50 °C) to give 6 as a brown oil that solidified upon standing. HPLC purity: ca. 81 area%; NMR content of ca. 57 wt%.
Compound 31 (26 g; 53 mmol; 1 eq.) was dissolved in ethanol (260 mL) and Noblyst Pl 155 (2.2 g; 10 % Pd; 54 wt% water) was added. The autoclave was flushed with nitrogen and hydrogen (5 bar) was added. The reaction mixture was stirred at room temperature for 32 hours. The reaction mixture was treated with charcoal (2 g), stirred for 15 min, and the charcoal was filtered off. The filtrate was concentrated via distillation and the residue (approximately 42 g) was diluted with heptane (200 mL). The mixture was heated to reflux to
obtain a clear solution. The solution was cooled to room temperature within 1 h and the resulting suspension was cooled to 0 °C and stirred for 2 hours at 0 °C. The solids were isolated via filtration and washed with heptane/ethanol (10:1; v/v; 3×10 mL). Compound 1 (18.0 g; 85 %) was dried under vacuum at 60 °C for 24 hours and obtained as a white, crystalline solid.
Compound 31 (26.5 kg; 53.5 mol; 1 eq.) was dissolved in ethanol (265 L) and Pd/C (2.0 kg; 10 % Pd; 54 wt% water) was added. The reactor was flushed with nitrogen, and hydrogen (4.5 bar) was added. The reaction mixture was stirred at 28-32 °C until the reaction was complete. The reaction mixture was treated with charcoal (1.3 kg) at a temperature of <
33 °C, stirred for 10 min, and the charcoal was filtered off, and the filter was washed with ethanol (10 L).The filtrates from two reactions were combined and concentrated via distillation under reduced pressure (max. 65 °C; distillate: min 480 L). The residue (approx. 50-60 L) was diluted with isopropylacetate (250 L). The mixture was again concentrated via distillation under reduced pressure (max. 65 °C; distillate: min 240-245 L). The residue (approx. 60-70 L) was cooled to 35-40 °C and isopropylacetate (125 L) and heptane (540 L) were added. The suspension was heated to reflux (approx. 88 °C) and stirred under reflux for 15-20 min. Subsequently, the mixture was cooled to 0-5 °C within 2 h and stirred at 0-5 °C for 2 hours. The solids were isolated via filtration and washed with heptane/isopropylacetate (5:1; v/v; 2×30 L; 0-5 °C). Wet 1 was dried under vacuum at 60 °C and was obtained as a white, crystalline solid (35.4 kg, 81.9%).
Preparation 3: Preparation of Compound 1 from Compound 6
At room temperature, 6 (3.00 g, 5.84 mmol) was dissolved in MeOH (19.8 mL). NaOH (1.0 M, 19.8 mL) was added in one portion and the reaction mixture was stirred for 1 h at room temperature. The reaction progress was monitored by HPLC, which showed 98% conversion after 1 h. Aq. HC1 (19.8 mL) was added and the mixture was diluted with water (120 mL) and MTBE (60 mL), resulting in a clear biphasic solution. After phase separation, the organic phase was washed with aq NaHC03 (20 mL). The organic layer was concentrated under high vacuum (25 mbar, 45 °C) to yield 2.77 g of 1 as a greenish oil. The identity was confirmed by comparison of HPLC retention time with an authentic sample of 1 as well as by 1H NMR.
Recrystallization of Compound 1
Wet 1 (40 kg; isopropylacetate/heptane wet) was treated with isopropylacetate (110 L) and heptane (440 L). The suspension was heated to reflux (approx. 88 °C) and stirred under reflux for 15-20 min. Subsequently, the mixture was cooled to 0-5 °C within 2 h and stirred at 0-5 °C for 2 hours. The solids were isolated via filtration and washed with
heptane/isopropylacetate (5:1; v/v; 2×30 L; 0-5 °C). A sample was taken for analysis
(criterion: a) purity; NLT 99.0 A% by HPLC; b) single impurities, NMT 0.15 A% by HPLC; c) enantiomer VT-463, NMT 1.0 A% by HPLC). Wet 1 was dried under vacuum at 60 °C for not less than 12 h. A sample was taken for analysis: criterion: a) LOD; NMT 0.5 wt% by gravimetry; b) residual toluene, NMT 890 ppm by HS-GC. 1 was obtained as a white, crystalline solid (28.5 kg, 66.7% from 31).
^ Jump up to:abRafferty SW, Eisner JR, Moore WR, Schotzinger RJ, Hoekstra WJ (2014). “Highly-selective 4-(1,2,3-triazole)-based P450c17a 17,20-lyase inhibitors”. Bioorg. Med. Chem. Lett. 24 (11): 2444–7. doi:10.1016/j.bmcl.2014.04.024. PMID24775307.
^ Jump up to:abcdToren PJ, Kim S, Pham S, Mangalji A, Adomat H, Guns ES, Zoubeidi A, Moore W, Gleave ME (2015). “Anticancer activity of a novel selective CYP17A1 inhibitor in preclinical models of castrate-resistant prostate cancer”. Mol. Cancer Ther. 14 (1): 59–69. doi:10.1158/1535-7163.MCT-14-0521. PMID25351916.
A Single arm, open label, signal seeking, Phase II a trial of the activity of seviteronel in patients with androgen receptor (AR) positive solid tumours
A Phase 2 Open-label Study to Evaluate the Efficacy and Safety of Seviteronel in Subjects With Castration-Resistant Prostate Cancer Progressing on Enzalutamide or Abiraterone
Innocrin Pharmaceuticals, Inc. Granted Fast Track Designation by FDA for VT-464 Treatment of Patients with Metastatic Castrate-resistant Prostate Cancer.
Innocrin Pharmaceuticals, Inc. Begins Phase 2 Study of Seviteronel in Women with Estrogen Receptor-positive or Triple-negative Breast Cancer and Expands Two Phase 2 Studies of Seviteronel in Men with Metastatic Castrate-resistant Prostate Cancer.
A Phase 2 Open-Label Study to Evaluate the Efficacy and Safety of VT-464 in Patients With Metastatic Castration Resistant Prostate Cancer Who Have Previously Been Treated With Enzalutamide, Androgen Receptor Positive Triple-Negative Breast Cancer Patients, and Men With ER Positive Breast Cancer
Innocrin Pharmaceuticals Inc. to Present Interim Results from Its Phase 1/2 Prostate Cancer Clinical Study and Preclinical Results That Demonstrate VT-464 Efficacy in a Clinically-Relevant Enzalutamide-Resistant Mouse Model.
A Phase 1/2 Open-Label Study to Evaluate the Safety, Pharmacokinetics, and Pharmacodynamics of Seviteronel in Subjects With Castration-Resistant Prostate Cancer
A Phase 1/2 Open-Label, Multiple-Dose Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Once-Daily VT-464 in Patients With Castration-Resistant Prostate Cancer
Innocrin Pharmaceuticals Appoints Fred Eshelman, PharmD as CEO and is Granted Fast Track Designation by FDA for Seviteronel Treatment of Women with Triple-negative Breast Cancer and Women or Men with Estrogen Receptor-positive Breast Cancer.
Gucalp A, Bardia A, Gabrail N, DaCosta N, Danso M, Elias AD, et al. Phase 1/2 study of oral seviteronel (VT-464), a dual CYP17-lyase inhibitor and androgen receptor (AR) antagonist, in patients with advanced AR positive triple negative (TNBC) or estrogen receptor (ER) positive breast cancer (BC). SABCS-2016 2016; abstr. P2-08-04.
Innocrin Pharmaceuticals Presents Data from the Ongoing Phase 2 Trial of Seviteronel in Estrogen Receptor-positive or Triple-negative Breast Cancer (CLARITY-01) at the San Antonio Breast Cancer Symposium.
Innocrin Pharmaceuticals, Inc. Appoints Edwina Baskin-Bey, MD as Chief Medical Officer and Expands the Ongoing Phase 2 Study of Seviteronel in Women with Estrogen Receptor-positive or Triple-negative Breast Cancer (TNBC).
A Phase 1/2 Open-Label Study to Evaluate the Safety, Pharmacokinetics, Pharmacodynamics and Efficacy of Seviteronel in Subjects With Advanced Breast Cancer
Speers CW, Chandler B, Zhao S, Liu M, Wilder-Romans K, Olsen E, et al. Radiosensitization of androgen receptor (AR)-positive triple-negative breast cancer (TNBC) cells using seviteronel (SEVI), a selective CYP17 lyase and AR inhibitor. ASCO-2017 2017; abstr. e12102.
GFH-018 , a TGFBR1 inhibitor, being investigated by GenFleet as an oral tablet formulation, for the treatment of cancer, including advanced solid tumors and hepatocellular carcinoma, in March 2019, the company was developing GFH-018 as a class 1 chemical drug in China, with a clinical trial expected to begin in the second half of 2019.
Transforming growth factor-β (TGF-β) is a multifunctional growth factor superfamily with extensive biological activity, involved in early embryonic development, cartilage and bone formation, extracellular matrix synthesis, inflammation, Interstitial fibrosis, regulation of immune and endocrine functions, tumor formation and development.
The TGF-β superfamily consists of a class of structural and functionally related polypeptide growth factors, including TGF-βs (ie, narrowly defined TGF-β), activins (axivins), inhibins, and bone morphogenetic proteins (BMPs). Müllerian inhibitors (mullerian), etc., TGF-β is one of the important members of this family. In mammals, TGF-β mainly exists in three forms of TGF-β1, TGF-β2 and TGF-β3, which are located on different chromosomes, and TGF-β1 accounts for the highest proportion (>90%) in somatic cells. It has the strongest activity, the most functions, and the widest distribution. The newly synthesized TGF-β appears as an inactive precursor consisting of a signal peptide, a latent-associated polypeptide (LAP) and a mature TGF-β. After enzymatic hydrolysis, it forms active TGF-β, and then Receptor binding exerts a biological effect.
TGF-[beta] signaling molecules signal through a transmembrane receptor complex. TGF-β receptor is a transmembrane protein present on the cell surface and is divided into type I receptor (TGF-βRI), type II receptor (TGF-βRII) and type III receptor (TGF-βRIII), of which TGF- βRI is also known as activin receptor-like kinase 5 (ALK5). TGF-βRIII lacks intrinsic activity and is primarily involved in the storage of TGF-β. TGF-βRI and TGF-βRII belong to the serine/threonine kinase family. Type II receptors bind to TGF-β ligands with higher affinity and form heterologous receptor complexes with type I receptors. Phosphorylation of a region rich in glycine and serine residues (GS domain) of the proximal membrane of the receptor initiates an intracellular signal cascade reaction.
Smads are important TGF-β signal transduction and regulatory molecules in cells, which can directly transduce TGF-β signaling from the cell membrane, such as the nucleus. TGF-β/Smads signaling pathway plays an important role in the occurrence and development of tumors. . In TGF-β/Smads signal transduction, activated TGF-β first binds to TGF-βRII on the cell membrane surface to form a heterodimeric complex, and TGF-βRI recognizes and binds to the binary complex.
TGF-βRII phosphorylates serine/threonine in the GS domain of the cytoplasmic domain of TGF-βRI, thereby activating TGF-βRI; activated TGF-βRI further phosphorylates R-Smads (Smad2/Smad3) protein, which in turn Co-Smad (Smad4) binds to a heterotrimeric complex that enters the nucleus and acts synergistically with other co-activators and co-inhibitors to regulate transcription of target genes. . Any change in any part of the TGF-β/Smads signaling pathway leads to abnormalities in the signal transduction pathway.
Current research indicates that in tumor cells, TGF-β can directly affect tumor growth (non-inherent effects of TGF-β signaling), or by inducing epithelial-mesenchymal transition, blocking anti-tumor immune responses, and increasing tumor-associated fibrosis And enhanced angiogenesis indirectly affects tumor growth (the intrinsic effect of TGF-β). At the same time, TGF-β has a strong fibrotic induction, which is an activator of tumor-associated fibroblasts. These fibroblasts are a major source of collagen type I and other fibrotic factors. Induction products of fibroblasts and other fibrotic factors may continue to develop a microenvironment that reduces immune responses, increases drug resistance, and enhances tumor angiogenesis. In addition, TGF-β affects blood vessels during individual development and tumor growth. Raw regeneration. For example, TGF-βRI-deficient mouse embryos show severe vascular development defects, demonstrating that the TGF-β signaling pathway is a key regulator in vascular endothelium and smooth muscle cell development.
In 2013, the FDA awarded Lilly’s small molecule TGF-βRI inhibitor LY2157299 (WO 2002/094833) for the treatment of glioma and liver cancer. LY2157299 is an orphan drug under research, named Galunisertib. Galunisertib inhibits tumor cell invasion and metastasis while inhibiting the infiltration of tumor cells into blood vessels. In the phase 2 clinical trial of patients with liver cancer, about 23% of patients treated with Galunisertib had a decrease in serum alpha-fetoprotein (AFP) levels of more than 20%. Compared with patients without AFP response, these patients had slower tumor progression and longer survival, and increased expression of cadherin in epithelial cells was also observed in these patients, suggesting that Galunisertib can be regulated by inhibiting TGF-β signaling pathway. EMT, thereby inhibiting the progression of liver cancer, the structure of Galunisertib (LY2157299) is shown in formula (II):
Background research and development materials refer to the following documents:
Step A: Ethyl acetate (291.41 ml, 2.98 mol) was dissolved in toluene (750.00 ml), and then sodium ethoxide (135.06 g, 1.98 mol) was added portionwise at room temperature, and the mixture was stirred at room temperature for 1 hour. Methyl 6-methylpyridine-2-carboxylate (150.00 g, 992.33 mmol) was added to the above reaction solution at 25 ° C, then heated to 95 ° C and stirred for 15 hours. The reaction mixture was cooled to 30 ° C, the pH was adjusted to 7 with acetic acid, diluted with water (500 ml), and ethyl acetate (500 ml). The organic phase was dried with anhydrous sodium s The residue was purified with EtOAc EtOAc EtOAc (EtOAc:EtOAc Rate: 58.35%).
Step B: Ethyl 3-(6-methyl-2-pyridine)-3-oxo-propanoate (120.00 g, 579.07 mmol) was dissolved in pyridine (300 mL) then 1-aminopyrrolidine- 2-keto-p-toluenesulfonate (172.01 g, 631.66 mmol). The reaction mixture was stirred at 25 ° C for 16 hours and then concentrated under reduced pressure to remove solvent. The residue was diluted with water (300 ml) and then extracted with ethyl acetate (300 ml). The combined organic phases were dried with anhydrous sodium s , yield: 90.28%).
Step C: Dissolving 3-(6-methyl-2-pyridine)-3-(2-carbonyl-pyrrolidine)imino-propionic acid ethyl ester (155.00 g, 535.72 mmol) in toluene and then adding ethanol Sodium (72.91 g, 1.07 mol). The reaction mixture was heated to 100 ° C and stirred for 16 hours, then cooled to room temperature. It was slowly diluted with water (1.5 liters), adjusted to pH 4 with concentrated hydrochloric acid, and extracted with dichloromethane / isopropyl alcohol (10/1) (1 liter x 7). The combined organic layers were dried with anhydrous sodium s The residue was triturated with petroleum ether / ethyl acetate = 10/1 (200 mL). The solid was dried under reduced pressure to give 2-(6-methyl-2-pyridine)-5,6-dihydro-4H-pyrrole[1,2-b]pyrazole-3-carboxylic acid (52.80 g, yield : 40.52%).
Step D: Dissolving 2-(6-methyl-2-pyridyl)-5,6-dihydro-4H-pyrrole[1,2-b]pyrazole-3-carboxylic acid (45.00 g, 184.99 mmol) In N,N-dimethylformamide (650.00 ml), then NBS (49.09 g, 258.99 mmol). The reaction mixture was stirred at 30-40 ° C for 60 hours, then diluted with water (600 mL) and extracted with dichloromethane / isopropyl alcohol (10/1) (500 mL × 3). The combined organic phases were washed with EtOAc (EtOAc m. The resulting solid was slurried with EtOAc/EtOAc =EtOAc (EtOAc). The solid was dried under reduced pressure to give 3-bromo-2-(6-methyl-2-pyridine)-5,6-dihydro-4H-pyrrole[1,2-b]pyrazole (33.00 g, yield: 64.13%).
Step E: 3-Bromo-2-(6-methyl-2-pyridyl)-5,6-dihydro-4H-pyrrole [1,2-b]pyrazole (1.00 g, 3.60 mmol) and boric acid Triisopropyl ester (1.79 g, 9.54 mmol) was dissolved in tetrahydrofuran (20.00 mL). The reaction mixture was cooled to minus 70 ° C, then n-butyllithium (2.5 M, 3.74 mL) was then added dropwise. After completion of the dropwise addition, the reaction mixture was stirred at 25 ° C for 1 hour, and then the pH was adjusted to 7 with aqueous hydrochloric acid (0.5 mol / liter). The tetrahydrofuran was then concentrated under reduced pressure and cooled to 15 °C. The mixture was filtered, and the filtered cake was purified with EtOAc EtOAc EtOAc (EtOAc) 5,6-Dihydro-4H-pyrrole[1,2-b]pyrazol-3-yl]boronic acid (750 mg, yield: 85.71%).
Preparation of Example 1:
Step A: 6-Iodo-[1,2,4]triazolo[1,5-a]pyridine (16.00 g, 65.30 mmol) was dissolved in tetrahydrofuran (800.00 mL) and cooled to below 60-70 ° C. Thereafter, lithium hexamethyldisilazide (1 mol/liter, 130.60 ml, 65.30 mmol) was added dropwise. The reaction mixture was stirred at minus 60-70 ° C for 15 minutes and N,N-dimethylformamide (14.32 g, 195.90 mmol, 15.07 mL). Stirring was then continued at minus 60 to 70 degrees C for 15 minutes and then quenched with saturated aqueous ammonium chloride (500 mL). The reaction mixture was warmed to room temperature and then extracted with ethyl acetate (500 ml). The combined organic layers were washed with EtOAc EtOAc m. The residue was purified with a silica gel column (eluent: methylene chloride / ethyl acetate = 10/1) to afford 6-iodo-[1,2,4]triazolo[1,5-a]pyridine-5- Formaldehyde (6.40 g, yield: 35.90%). . 1H NMR (400 MHz, DMSO-d6) 10.46 (S, IH), 8.62 (S, IH), 8.16 (D, J = 9.3Hz, IH), 7.88 (D, J = 9.3Hz, IH).
Step B: To a 500 ml three-necked flask equipped with a thermometer and a nitrogen balloon, 2-diethoxyphosphorylacetonitrile (3.83 g, 21.61 mmol, 3.48 ml) and tetrahydrofuran (80 ml) were added. The mixture was cooled to 0.degree. C. and then potassium tert-butoxide (2.42 g, 21.61 mmol). The reaction mixture was stirred at 0 ° C for 15 minutes and then added dropwise to another suspension through a dropping funnel (dispersing 6-iodo-[1,2,4]triazolo[1,5-a]pyridine-5-carbaldehyde In tetrahydrofuran (120 ml) and cooled to 0 ° C). The reaction mixture was stirred at 0<0>C for 15 min then EtOAc (EtOAc)EtOAc. The combined organic layers were washed with EtOAc EtOAc m. The residue was purified with a silica gel column (eluent: methylene chloride / ethyl acetate = 200/1 to 10/1) to afford (E)-3-(6-iodo-[1,2,4]triazole. [1,5-a]pyridin-5-yl)prop-2-enenitrile (4.2 g, yield: 65.66%). . 1 H NMR (400 MHz, CHLOROFORM-D) [delta] 8.42 (S, IH), 8.03 (D, J = 9.3Hz, IH), 7.98-7.91 (m, IH), 7.85-7.78 (m, IH), 7.60 (d, J = 9.2 Hz, 1H).
Step C: (E)-3-(6-Iodo-[1,2,4]triazolo[1,5-a]pyridin-5-yl)prop-2-enenitrile (4.50 g, 15.20 m Mole), [2-(6-methyl-2-pyridyl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl]boronic acid (4.43 g, 18.24 m Mole), sodium carbonate (4.83 g, 45.60 mmol), [1,1′-bis(diphenylphosphino)ferrocene]palladium dichloride (556.07 mg, 759.96 μmol), 2-dicyclohexylphosphine- 2′,6′-dimethoxybiphenyl (311.98 mg, 759.96 μmol) and [2-(2-aminophenyl)phenyl]-chloro-palladium-cyclohexyl-[2-(2,6- Dimethoxyphenyl)phenyl]phosphine (547.64 mg, 759.96 μmol) was added to a mixed solvent of dioxane (100 ml) and water (20 ml). It was replaced with nitrogen 3 times and then heated to 90-100 ° C and stirred for 2 hours. The reaction mixture was poured into water (200 ml) and evaporated and evaporated. The combined organic layers were washed with EtOAc EtOAc m. The residue was purified with EtOAc mjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjj The solid was concentrated and dried under reduced pressure to give (E)-3-[6-[2-(6-methyl-2-pyridyl)-5,6-dihydro-4H-pyrrolo[1,2-b] Pyrazol-3-yl]-[1,2,4]triazolo[1,5-a]pyridin-5-yl]prop-2-enenitrile (5.37 g, yield: 96.16%). . 1 H NMR (400 MHz, CHLOROFORM-D) [delta] 8.49 (S, IH), 7.82-7.74 (m, 2H), 7.59-7.46 (m, 4H), 6.99 (dd, J = 2.6,6.1Hz, IH) , 4.39 (d, J = 6.3 Hz, 2H), 2.90 – 2.70 (m, 4H), 2.20 (s, 3H).
Step D: (E)-3-[6-[2-(6-Methyl-2-pyridyl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole-3 -yl]-[1,2,4]triazolo[1,5-a]pyridin-5-yl]prop-2-enenitrile (5.37 g, 14.62 mmol) dissolved in dichloromethane (20 mL) , a mixed solvent of dimethyl sulfoxide (70 ml) and water (20 ml), then separately added hydrogen peroxide (8.29 g 73.10 mmol, 7.02 ml, 30%) and sodium hydroxide (2 mol / liter, 14.62 ml) ). The mixture was stirred at 15-20 degrees Celsius for 12 hours. The mixture was poured into water (200 ml), and extracted with a mixture solvent of dichloromethane/isopropanol (3/1) (200 ml × 1). The organic layer was washed with EtOAc EtOAc m. The residue was purified by preparative high performance liquid chromatography (column: Phenomenex Gemini C18 250 x 50 mm x 10 μm; mobile phase: [water (0.05% ammonia v/v)-acetonitrile]; gradient: 5%-32%, 33 80% min) Example 1 (3.6 g, yield: 63.82%) was obtained. . 1 H NMR (400 MHz, CHLOROFORM-D) [delta] 8.45 (S, IH), 8.09 (D, J = 15.6Hz, IH), 7.85 (D, J = 15.6Hz, IH), 7.69 (D, J = 9.2 Hz, 1H), 7.55-7.45 (m, 2H), 7.37 (d, J = 7.8 Hz, 1H), 6.99 (d, J = 7.7 Hz, 1H), 5.93-5.65 (m, 2H), 4.35 (br .s., 2H), 2.99-2.64 (m, 4H), 2.33 (s, 3H).
Novel crystalline and salt (hydrochloride, sulfate and mesylate) forms of a TGF-βRI inhibitor, designated as Forms A and B, processes for their preparation and compositions comprising them are claimed for treating cancers. The compound was originally claimed in WO2017215506 , assigned to Medshine Discovery Inc alone.
Example 1 Preparation of a compound of formula (I)
Preparation of intermediates 1-6:
Step A: Ethyl acetate (291.41 ml, 2.98 mol) was dissolved in toluene (750.00 ml), and then sodium ethoxide (135.06 g, 1.98 mol) was added portionwise at room temperature, and the mixture was stirred at room temperature for 1 hour. 1-1 (150.00 g, 992.33 mmol) was added to the above reaction liquid at 25 ° C, and then heated to 95 ° C and stirred for 15 hours. The reaction mixture was cooled to about 30 ° C, and the pH was adjusted to 7 with acetic acid, diluted with water (500 ml), and ethyl acetate (500 ml). The organic phase was dried with anhydrous sodium s The residue was purified with a silica gel column (eluent: petroleum ether/ethyl acetate v/v = 50/1) to afford 1-2.
Step B: Dissolve 1-2 (120.00 g, 579.07 mmol) in pyridine (300 mL), then add 1-aminopyrrolidin-2-one p-toluenesulfonate (172.01 g, 631.66 mmol) ). The reaction mixture was stirred at 25 ° C for 16 hours and then concentrated under reduced vacuo. The residue was diluted with water (300 ml) and then extracted with ethyl acetate (300 ml). The combined organic layers were dried with anhydrous sodium s
Step C: 1-3 (155.00 g, 535.72 mmol) was dissolved in toluene then sodium ethoxide (72.91 g, 1.07 mol). The reaction mixture was heated to 100 ° C and stirred for 16 hours, then cooled to room temperature. It was slowly diluted with water (1.5 liters), adjusted to pH 4 with concentrated hydrochloric acid, and extracted with dichloromethane/isopropanol (v/v = 10/1, 1 liter x 7). The combined organic layers were dried with anhydrous sodium s The residue was triturated with petroleum ether / ethyl acetate (v/v = 10/1, 200 mL). The solid was dried under reduced pressure to give 1-4.
Step D: 1-4 (45.00 g, 184.99 mmol) was dissolved in N,N-dimethylformamide (650.00 ml), then NBS (49.09 g, 258.99 mmol). The reaction mixture was stirred at 30 to 40 ° C for 60 hours, then diluted with water (600 ml), and extracted with dichloromethane / isopropyl alcohol (v / v = 10 / 1,500 ml × 3). The combined organic phases were washed with EtOAc (EtOAc m. The resulting solid was slurried with EtOAc/EtOAc (EtOAc/EtOAc) The solid was dried under reduced pressure to give 1-5.
Step E: 1-5 (1.00 g, 3.60 mmol) and triisopropyl borate (1.79 g, 9.54 mmol) were dissolved in tetrahydrofuran (20.00 mL). The reaction mixture was cooled to minus 70 ° C, then n-butyllithium (2.5 M, 3.74 mL) was added dropwise. After completion of the dropwise addition, the reaction mixture was stirred at 25 ° C for 1 hour, and then the pH was adjusted to 7 with aqueous hydrochloric acid (0.5 mol / liter). It was then concentrated under reduced pressure to remove tetrahydrofuran and cooled to 15 °C. The mixture was filtered, and the EtOAc EtOAc m.
Preparation of the compound of formula (I):
Step A: 1-7 (16.00 g, 65.30 mmol) was dissolved in tetrahydrofuran (800.00 ml), cooled to minus 60-70 ° C, and lithium hexamethyldisilazide (1 mol/L, 130.60) was added dropwise. ML, 65.30 mmol). The reaction mixture was stirred at -60 to 70 ° C for 15 minutes, and N,N-dimethylformamide (14.32 g, 195.90 mmol, 15.07 ml) was added. Stirring was then continued at minus 60-70 ° C for 15 minutes and then quenched with saturated aqueous ammonium chloride (500 mL). The reaction mixture was warmed to room temperature and then extracted with ethyl acetate (500 ml). The combined organic layers were washed with EtOAc EtOAc m. The residue was purified with a silica gel column (eluent: methylene chloride / ethyl acetate v/v = 10/1) to afford 1-8. . 1 H NMR (400 MHz, DMSO-d6) 10.46 (S, IH), 8.62 (S, IH), 8.16 (D, J = 9.3Hz, IH), 7.88 (D, J = 9.3Hz, IH).
Step B: To a 500 ml three-necked flask equipped with a thermometer and a nitrogen balloon, 2-diethoxyphosphorylacetonitrile (3.83 g, 21.61 mmol, 3.48 ml) and tetrahydrofuran (80 ml) were added. The mixture was cooled to 0 ° C then potassium tert-butoxide (2.42 g, 21.61 mmol). The reaction mixture was stirred at 0<0>C for 15 min then added dropwise to a further suspension (1~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~ The reaction mixture was stirred at 0<0>C for 15 min then EtOAc (EtOAc)EtOAc. The combined organic layers were washed with EtOAc EtOAc m. The residue was purified with a silica gel column (eluent: methylene chloride/ethyl acetate v/v = 200/1 to 10/1) to afford 1-9. . 1 H NMR (400 MHz, CDCl3 . 3 ) [delta] 8.42 (S, IH), 8.03 (D, J = 9.3Hz, IH), 7.98-7.91 (m, IH), 7.85-7.78 (m, IH), 7.60 ( d, J = 9.2 Hz, 1H).
Step C: 1-9 (4.50 g, 15.20 mmol), 1-6 (4.43 g, 18.24 mmol), sodium carbonate (4.83 g, 45.60 mmol), [1,1′-bis (diphenyl) Phosphine) ferrocene] palladium dichloride (556.07 mg, 759.96 μmol), 2-biscyclohexylphosphine-2′, 6′-dimethoxybiphenyl (311.98 mg, 759.96 μmol) and [2-( 2-Aminophenyl)phenyl]-chloro-palladium-cyclohexyl-[2-(2,6-dimethoxyphenyl)phenyl]phosphine (547.64 mg, 759.96 μmol) was added to the dioxane (100 ml) and water (20 ml) in a mixed solvent. It was replaced with nitrogen three times and then heated to 90 to 100 ° C and stirred for 2 hours. The reaction mixture was poured into water (200 ml) and evaporated and evaporated. The combined organic layers were washed with EtOAc EtOAc m. The residue was purified on a silica gel column (eluent: methylene chloride/methanol, v/v=30/1) to afford crude crude product in petroleum ether/ethyl acetate (v/v=5/1) After stirring for 12 hours, the solid was collected by filtration, and the solid was concentrated and dried under reduced pressure to give 1-10. . 1 H NMR (400 MHz, CDCl3 . 3 ) [delta] 8.49 (S, IH), 7.82-7.74 (m, 2H), 7.59-7.46 (m, 4H), 6.99 (dd, J = 2.6,6.1Hz, IH), 4.39 (d, J = 6.3 Hz, 2H), 2.90 – 2.70 (m, 4H), 2.20 (s, 3H).
Step D: 1-10 (5.37 g, 14.62 mmol) was dissolved in a mixed solvent of dichloromethane (20 ml), dimethyl sulfoxide (70 ml) and water (20 ml), and then hydrogen peroxide ( 8.29 g 73.10 mmol, 7.02 mL, 30%) and sodium hydroxide (2 mol/L, 14.62 mL). The mixture was stirred at 15 to 20 ° C for 12 hours. The mixture was poured into water (200 ml), and extracted with a mixture solvent of dichloromethane/isopropanol (3/1) (200 ml × 1). The organic layer was washed with EtOAc EtOAc m. The residue was purified by preparative high performance liquid chromatography (column: Phenomenex Gemini C18 250 x 50 mm x 10 μm; mobile phase: [water (0.05% ammonia v/v)-acetonitrile]; gradient: 5%-32%, 33 80% minute) to give a compound of formula (I). . 1 H NMR (400 MHz, CDCl3 . 3 ) [delta] 8.45 (S, IH), 8.09 (D, J = 15.6Hz, IH), 7.85 (D, J = 15.6Hz, IH), 7.69 (D, J = 9.2Hz , 1H), 7.55-7.45 (m, 2H), 7.37 (d, J = 7.8 Hz, 1H), 6.99 (d, J = 7.7 Hz, 1H), 5.93-5.65 (m, 2H), 4.35 (br. s., 2H), 2.99-2.64 (m, 4H), 2.33 (s, 3H).
Example 2 Preparation of a compound of formula (II)
115 mg of the compound of formula (I) was added to an 8 ml glass vial, 4 ml of tetrahydrofuran was added, and the solution was sonicated by ultrasonication; then 1.05 equivalent of p-toluenesulfonic acid monohydrate was slowly added. The suspension sample was placed on a magnetic stirrer (40 ° C) and stirred for 16 hours. The sample solution was centrifuged, and the solid was taken out and dried in a vacuum oven at 35 ° C for 16 hours to obtain a compound of the formula (II). 1 H NMR (400 MHz, CD 3 OD) δ 8.61 (s, 1H), 8.14 (t, J = 8.0 Hz, 1H), 8.05 (d, J = 15.6 Hz, 1H), 7.90 (d, J = 8.8 Hz, 1H), 7.70 (dd, J=8.4, 15.6 Hz, 4H), 7.54 (d, J = 15.6 Hz, 1H), 7.39 (d, J = 8.0 Hz, 1H), 7.20 (d, J = 7.6) Hz, 2H), 4.42 (m, 2H), 3.05-2.87 (m, 2H), 2.82 (s, 3H), 2.81-2.74 (m, 2H), 2.35 (s, 3H).
Example 3 Preparation of a compound of formula (IV)
115 mg of the compound of formula (I) was added to an 8 ml glass vial, 4 ml of tetrahydrofuran was added, and the solution was sonicated by ultrasonication; then 1.05 equivalent of hydrochloric acid was slowly added. The suspension sample was placed on a magnetic stirrer (40 ° C) and stirred for 16 hours. The sample solution was centrifuged, and the solid was taken out and dried in a vacuum oven at 35 ° C for 16 hours. The obtained solid was added to an appropriate amount of acetone to prepare a suspension and stirred at 40 ° C, and the supernatant was discarded by centrifugation, and the solid sample was drained with an oil pump at room temperature to obtain a compound of the formula (IV).
Example 4 Preparation of a compound of formula (V)
115 mg of the compound of formula (I) was added to an 8 ml glass vial, 4 ml of tetrahydrofuran was added, and the solution was sonicated by ultrasonication; then 1.05 equivalent of sulfuric acid was slowly added. The suspension sample was placed on a magnetic stirrer (40 ° C) and stirred for 16 hours. The sample solution was centrifuged, and the solid was taken out and dried in a vacuum oven at 35 ° C for 16 hours to obtain a compound of the formula (V).
Example 5 Preparation of a compound of formula (VI)
115 mg of the compound of formula (I) was added to an 8 ml glass vial, 4 ml of tetrahydrofuran was added, and the solution was sonicated by ultrasonication; then 1.05 equivalent of methanesulfonic acid was slowly added. The suspension sample was placed on a magnetic stirrer (40 ° C) and stirred for 16 hours. The sample solution was centrifuged, and the solid was taken out and dried in a vacuum oven at 35 ° C for 16 hours to obtain a compound of the formula (VI).
Example 6 Preparation of Form A of Compound of Formula (I)
10 g of the compound of the formula (I) was placed in a mixed solvent of ethanol (80 ml) and water (40 ml), heated to 70-75 ° C and stirred until clarified, and filtered while hot, and the filtrate was distilled under reduced pressure to a volume of the remaining solution. 50 ml, followed by cooling to stand for crystallisation, filtration, and the resulting filter cake was dried under reduced pressure to give a solid of the compound of formula (I).
Example 7 Preparation of Form B of Compound of Formula (II)
192 mg of the compound of formula (I) was weighed into a glass bottle. 10 ml of a tetrahydrofuran:acetic acid (v/v=9/1) mixed solvent was added, and after ultrasonic assisted for 30 minutes, the sample was dissolved into a clear solution. Stir on a magnetic stirrer (40 ° C). After 1.05 equivalents of p-toluenesulfonic acid monohydrate was slowly added, the sample was stirred overnight. After naturally cooling to room temperature, the supernatant was discarded by centrifugation, stirred for 10 hours by adding 10 ml of tetrahydrofuran, and the supernatant was discarded by centrifugation, and the same procedure was repeated twice more. The obtained solid was dried in a vacuum oven at 40 ° C for 1 hour, and after milling, it was further dried in a vacuum oven at 30 ° C for 16 hours to obtain a crystal form B of the compound of the formula (II).
Maleate in anhydrous or monohydrate CAS, 2326561-36-6, AND 2326561-38-8 form are BTK and HER-2 kinase inhibitor useful for treating cancer
Useful for treating breast cancer, ovary cancer and colon cancer. are BTK and HER-2 kinase inhibitor useful for treating cancer.
Anticancer protein kinase inhibitor
The compound was originally claimed in WO2013152135 , and may provide the structure of TL-487 , a small molecule inhibitor to HERs, being investigated by Teligene for the treatment of breast cancer; in July 2016, the company intended to develop the product as a class 1.1 chemical drug in China.
Novel crystalline maleate salt of (E)-N-(3-cyano-7-ethoxy-4-((4-phenoxyphenyl)amino)quinolin-6-yl)-4-(dimethylamino)but-2-enamide (first disclosed in WO2013152135) and its hydrates (monohydrate) and anhydrates, process for its preparation, composition comprising it and its use for treating cancers such as breast cancer, ovary cancer, colon cancer, prostate cancer, kidney cancer, bladder cancer, stomach cancer, lung cancer, mantle cell lymphoma and multiple myeloma are claimed. The compound is disclosed to be an irreversible inhibitor to BTK and Her-2 (also known as Erb-2 or neu).
(E) -N- (3-cyano-7-ethoxy-4- ( (4-phenoxyphenyl) amino) quinolin-6-yl) -4- (dimethylamino) but-2-enamide is mentioned in WO2013152135 and corresponds to the compound of the Formula I:
Formula I
Compounds derived from 3-cyanoquinoline have been shown to have anti-tumor activity, which may make them useful as chemotherapeutic agents in treating various cancers, including but not limited to, pancreatic cancer, melanoma, lymphatic cancer, parotid tumors, Barrett’s esophagus, esophageal carcinomas, head and neck tumors, ovarian cancer, breast cancer, epidermoid tumors, cancers of major organs, such as kidney, bladder, larynx, stomach, and lung, colonic polyps and colorectal cancer and prostate cancer. Examples of compounds derived from 3-cyanoquinoline are disclosed and shown to possess anti-tumor activity in many literatures. One limitation of certain 3-cyanoquinoline compounds is that they are not water soluble in a free base form.
The crystalline form of a particular drug as a salt, a hydrate and/or any polymorph thereof is often one important determinant of the drug’s ease of preparation, stability, water solubility, storage stability, ease of formulation and in-vivo pharmacology. It is possible that one crystalline form is preferable over another where certain aspects such as ease of preparation, stability, water solubility and/or superior pharmacokinetics are deemed to be critical. Crystalline forms of (E) -N- (3-cyano-7-ethoxy-4- ( (4-phenoxyphenyl) amino) quinolin-6-yl) -4- (dimethylamino) but-2-enamide salts that possess a higher degree of water solubility than the free base but are stable fulfill an unmet need for stable, crystalline, water-solubl
95%ethanol (4.0 ml) was added to (E) -N- (3-cyano-7-ethoxy-4- ( (4-phenoxyphenyl) amino) quinolin-6-yl) -4- (dimethylamino) but-2-enamide (500 mg, 0.99 mmol, 1.0 eq) , followed sulfuric acid (101.9 mg, 1.04 mmol, 1.05 eq) in 95%ethanol (1.0 ml) was added dropwise to the reaction mixture. Then an amount of precipitate was founded. Another 95% (60 ml) was added to the reaction mixture and the reaction mixture was heated to 70℃. Filtered and the filtrate was heated to 70℃ again. Then the reaction mixture was cooled to room temperature and The reaction mixture was crystallized at -10℃ for 41.5h. Filtered the precipitated solid and dried at 40℃ under vacuum for 1 hour to get the title compound (260 mg) as a yellow solid.
X-ray detection shows an amorphous structure to the compound as FIG. 9.
Example 2. Synthesis of (E) -N- (3-cyano-7-ethoxy-4- ( (4-phenoxyphenyl) amino) quinolin-6-yl) -4- (dimethylamino) but-2-enamide hydrochloride
95%ethanol (5.0 ml) was added to (E) -N- (3-cyano-7-ethoxy-4- ( (4-phenoxyphenyl) amino) quinolin-6-yl) -4- (dimethylamino) but-2-enamide (500 mg, 0.99 mmol, 1.0 eq) , followed hydrochloric acid (38.0 mg, 1.04 mmol, 1.05 eq) in 95%ethanol (1.0 ml) was added dropwise to the reaction mixture. The reaction mixture was heated to 70℃. Filtered and the filtrate was crystallized under -10℃ for 44.5h. Filtered the precipitated solid and dried at 40℃ under vacuum for 1 hour to get the title compound (96 mg) as a yellow solid.
X-ray detection shows an amorphous structure to the compound in FIG. 6.
Example 3. Synthesis of (E) -N- (3-cyano-7-ethoxy-4- ( (4-phenoxyphenyl) amino) quinolin-6-yl) -4- (dimethylamino) but-2-enamide malate
(E) -N- (3-cyano-7-ethoxy-4- ( (4-phenoxyphenyl) amino) quinolin-6-yl) -4- (dimethylamino) but-2-enamide (500 mg, 0.99 mmol, 1.0 eq) , L-malic acid (139.4 mg, 1.04 mmol, 1.05 eq) and 95%ethanol (5.0 ml) was added to a 50 ml round-bottom flask. The reaction mixture was heated to 70℃. Filtered and the filtrate was crystallized under -10℃ for 45.5h. A little of precipitate was founded and then the reaction mixture was evaporated under vacuum at 40℃ to give the target (370 mg) as a yellow solid.
X-ray detection shows an amorphous structure to the compound in FIG. 8
Example 4: synthesis of (E) -N- (3-cyano-7-ethoxy-4- ( (4-phenoxyphenyl) amino) quinolin-6-yl) -4- (dimethylamino) but-2-enamide citrate
To a solution of (E) -N- (3-cyano-7-ethoxy-4- ( (4-phenoxyphenyl) amino) quinolin-6-yl) -4- (dimethylamino) but-2-enamide (500 mg, 0.99 mmol, 1.0 eq) , citric acid (198.8 mg, 1.04 mmol, 1.05 eq) and 95%ethanol (5.0 ml) . The reaction mixture was heated to 70℃. Filtered and the filtrate was crystallized under -10℃ for 45h. A little of precipitate was founded and then the reaction mixture was evaporated under vacuum at 40℃ to give the target compound (610 mg) as a yellow solid.
X-ray detection shows an crystalline structure to the compound in FIG. 7.
Example 5: Preparation of (E) -N- (3-cyano-7-ethoxy-4- ( (4-phenoxyphenyl) amino) quinolin-6-yl) -4- (dimethylamino) but-2-enamide maleate monohydrate.
(E) -N- (3-cyano-7-ethoxy-4- ( (4-phenoxyphenyl) amino) quinolin-6-yl) -4- (dimethylamino) but-2-enamide free base (0.091 kg) is rinsed with a 10%solution of USP purified water in n-propanol (0.082 kg, 0.10 L) followed by the addition of water: n-propanol solution (0.74 kg, 0.90 L) . Maleic acid is added (1.01 equiv) and the mixture is rinsed with 10%water: n-propanol (0.082 kg, 0.10 L) . The mixture is quickly heated to 50-60 ℃ and held for a minimum of 15 min. until a solution is obtained. The hot solution is clarified through a pre-heated 50-60 ℃, 0.2 Mm filter cartridge and the filtrates are collected in a preheated 45-55℃, 2 L multi-neck flask. The filter cartridge is rinsed through with 10%water: n-propanol pre-heated to 45-55 ℃ (0.082 kg, 0.10 L) . The solution is cooled over at least one hour to 40 ℃ and held at that temperature for 12 hours then cooled to room temperature (25 ℃) over a minimum of four hours and held at that temperature for at least two hours. The mixture is filtered on a 12.5 cm diameter Buchner funnel for 5 min., then rinsed and washed with prefiltered10%water: n-propanol solution (2 x 0.12 kg, 2 x 0.15 L) . The cake is dammed and suction maintained until dripping essentially stops, about 1 h.
PXRD is shown in FIG. 1.
Example 6: The product from Example 1 is dried (50 ℃, 10 mm Hg, 24 h) to give crystalline, anhydrous (E) -N- (3-cyano-7-ethoxy-4- ( (4-phenoxyphenyl) amino) quinolin-6-yl) -4- (dimethylamino) but-2-enamide maleate.
PXRD is shown in FIG. 3.
Example 7: Preparation of (E) -N- (3-cyano-7-ethoxy-4- ( (4-phenoxyphenyl) amino) quinolin-6-yl) -4- (dimethylamino) but-2-enamide maleate monohydrate.
To a solution of (E) -N- (3-cyano-7-ethoxy-4- ( (4-phenoxyphenyl) amino) quinolin-6-yl) -4- (dimethylamino) but-2-enamide (38.0 g, 75.0 mmol, 1.0 eq) and n-propanol/H 2O (380 ml, V: V=9: 1) . maleic acid (8.7 g, 75.0 mmol, 1.0 eq) in n-propanol/H 2O (76 ml, V: V=9: 1) was added to the reaction mixture. An amount of precipitate was founded, then the reaction mixturewas heated to 65 ℃. The solid was dissolved completely, then the reaction mixture was cooled to room temperature and stand for 20 hours. Filtered and filtrate was evaporated under vacuum to get the crude product.
The crude product (14.0 g) was recrystallized in n-propanol/H 2O (240 ml, V: V=9: 1) at 70℃. The solid was dissolved completely, then the reaction mixture was cooled to room temperature and stand for 20.5 hours. Filtered and wash the cake with n-propanol/H 2O (20 ml, V: V=9: 1) to get target product (12.9 g, wet) .
To a solution of (E) -N- (3-cyano-7-ethoxy-4- ( (4-phenoxyphenyl) amino) quinolin-6-yl) -4- (dimethylamino) but-2-enamide (21.5 g, 42.4 mmol, 1.0 eq) and ethanol (300 ml) . maleic acid (5.2 g, 44.8 mmol, 1.05 eq) was added to the reaction mixture. An amount of precipitate was founded, then the reaction mixture was heated to 70 ℃. Another ethanol (1980 ml) was added to the reaction mixture in several times and the reaction temperature was keep at 70 ℃. Filtered and filtrate was cooled to room temperature, stop stirring and stand for 16-20 hours. Filtered and the solid was dried at room temperature for 24 hours to get the title compound.
Being investigated by Jiangsu Hansoh, Shanghai Hansoh Biomedical and Changzhou Hengbang Pharmaceutical ; in June 2018, the product was being developed as a class 1 chemical drug in China.
Useful for treating liver cancer, gastric cancer and prostate cancer.
Use for treating cancers, liver cancer, gastric cancer, prostate cancer, skin cancer, ovary cancer, lung cancer, breast cancer, colon cancer, glioma and rhabdomyosarcoma
The fibroblast growth factor receptor (FGFR) belongs to the receptor tyrosine kinase transmembrane receptor and includes four receptor subtypes, namely FGFR1, FGFR2, FGFR3 and FGFR4. FGFR regulates various functions such as cell proliferation, survival, differentiation and migration, and plays an important role in human development and adult body functions. FGFR is abnormal in a variety of human tumors, including gene amplification, mutation and overexpression, and is an important target for tumor-targeted therapeutic research.
FGFR4, a member of the FGFR receptor family, forms dimers on the cell membrane by binding to its ligand, fibroblast growth factor 19 (FGF19), and the formation of these dimers can cause critical tyrosine in FGFR4’s own cells. The phosphorylation of the amino acid residue activates multiple downstream signaling pathways in the cell, and these intracellular signaling pathways play an important role in cell proliferation, survival, and anti-apoptosis. FGFR4 is overexpressed in many cancers and is a predictor of malignant invasion of tumors. Decreasing and reducing FGFR4 expression can reduce cell proliferation and promote apoptosis. Recently, more and more studies have shown that about one-third of liver cancer patients with continuous activation of FGF19/FGFR4 signaling pathway are the main carcinogenic factors leading to liver cancer in this part of patients. At the same time, FGFR4 expression or high expression is also closely related to many other tumors, such as gastric cancer, prostate cancer, skin cancer, ovarian cancer, lung cancer, breast cancer, colon cancer and the like.
The incidence of liver cancer ranks first in the world in China, with new and dead patients accounting for about half of the total number of liver cancers worldwide each year. At present, the incidence of liver cancer in China is about 28.7/100,000. In 2012, there were 394,770 new cases, which became the third most serious malignant tumor after gastric cancer and lung cancer. The onset of primary liver cancer is a multi-factor, multi-step complex process with strong invasiveness and poor prognosis. Surgical treatments such as hepatectomy and liver transplantation can improve the survival rate of some patients, but only limited patients can undergo surgery, and most patients have a poor prognosis due to recurrence and metastasis after surgery. Sorafenib is the only liver cancer treatment drug approved on the market. It can only prolong the overall survival period of about 3 months, and the treatment effect is not satisfactory. Therefore, it is urgent to develop a liver cancer system treatment drug targeting new molecules. FGFR4 is a major carcinogenic factor in liver cancer, and its development of small molecule inhibitors has great clinical application potential.
At present, some FGFR inhibitors have entered the clinical research stage as anti-tumor drugs, but these are mainly inhibitors of FGFR1, 2 and 3, and the inhibition of FGFR4 activity is weak, and the inhibition of FGFR1-3 has hyperphosphatemia. Such as target related side effects. Highly selective inhibitor of FGFR4 can effectively treat cancer diseases caused by abnormal FGFR4 signaling pathway, and can avoid the side effects of hyperphosphatemia caused by FGFR1-3 inhibition. Highly selective small molecule inhibitors against FGFR4 in tumor targeted therapy The field has significant application prospects.
SYN
PATENT
WO2017198149
where it is claimed to be an FGFR-4 inhibitor for treating liver and prostate cancers, assigned to Jiangsu Hansoh Pharmaceutical Group Co Ltd and Shanghai Hansoh Biomedical Co Ltd .
PATENT
WO2019085860
Compound (R)-N-(5-Cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-formyl-6-((2-carbonyl-) 1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide (shown as Formula I). The compound of formula (I) is disclosed in Hausen Patent PCT/CN2017/084564, the compound of formula I is a fibroblast growth factor receptor inhibitor, and the fibroblast growth factor receptor (FGFR) belongs to the receptor tyrosine kinase transmembrane receptor. The body includes four receptor subtypes, namely FGFR1, FGFR2, FGFR3 and FGFR4. FGFR regulates various functions such as cell proliferation, survival, differentiation and migration, and plays an important role in human development and adult body functions. FGFR is abnormal in a variety of human tumors, including gene amplification, mutation and overexpression, and is an important target for tumor-targeted therapeutic research.
Example 1: Preparation of a compound of formula (I)
[0048]
First step 4-(((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)amino)butane Preparation of 1-propanol
[0049]
[0050]
2-(Dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-carbaldehyde (1.0 g, 4.2 mmol), 4-aminobutyl at room temperature l-ol (0.45g, 5.1mmol) was dissolved in DCE (15mL), stirred for 2 hours, followed by addition of NaBH (OAc) . 3 (1.35 g of, 6.4 mmol), stirred at room temperature overnight. The reaction was treated with CH 2 CI 2 was diluted (100 mL), the organic phase was washed with water (10mL) and saturated brine (15mL), and dried over anhydrous sodium sulfate, and concentrated by column chromatography to give compound 4 – (((2- ( Dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)amino)butan-1-ol (0.9 g, 69%) .
The second step is 3-((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)-1,3- Preparation of oxazepine-2 ketone
[0054]
[0055]
4-(((2-(Dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)amino) in an ice water bath Butan-1-ol (0.6 g, 1.94 mmol) was dissolved in DCE (15 mL), then bis(trichloromethyl) carbonate (0.22 g, 0.76 mmol) was added and triethylamine (0.78 g, 7.76) was slowly added dropwise. Methyl) and then stirred at room temperature for 3 hours. The reaction temperature was raised to 80 ° C, and the reaction was carried out at 80 ° C for 6 hours. After the reaction was cooled to room temperature, it was diluted with CH 2 Cl 2 (100 mL), and the organic phase was washed sequentially with water (10 mL) and brine (15 mL) Drying with sodium sulfate, concentration and column chromatography to give the compound 3-((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl) )methyl)-1,3-oxazepin-2-one (0.37 g, 57%).
[0056]
MS m/z (ESI): 336.2 [M+H] + .
[0057]
The third step is phenyl 7-(dimethoxymethyl)-6-((2-carbonyl-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1, Preparation of 8-naphthyridin-1(2H)-carboxylate
[0058]
[0059]
3-((2-(Dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)-1,3-oxan -2-one (670mg, 2mmol), diphenyl carbonate (643mg, 3mmol) mixing in of THF (15 mL), N 2 in an atmosphere, cooled to -78 deg.] C, was added dropwise LiHMDS in THF (4mL, 4mmol) was Naturally, it was allowed to react to room temperature overnight. After adding saturated aqueous NH 4 Cl (100 mL), ethyl acetate (100 mL×2), EtOAc. Methyl)-6-((3-carbonylmorpholino)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxylate (432 mg, 47%) .
The fourth step: (R)-N-(5-cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-(dimethoxymethyl) -6-((2-carbonyl-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide synthesis
[0063]
[0064]
(R)-6-Amino-4-((1-methoxypropan-2-yl)amino) nicotinenitrile (30 mg, 0.14 mmol), phenyl 7-(dimethoxymethyl)-6- ( (2-carbonyl-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxylate (60 mg, 0.13 Methyl acetate was dissolved in THF (5 mL), cooled to -78 ° C under N 2atmosphere, and a solution of THF (0.3 mL, 0.3 mmol) of LiHMDS was added dropwise to the reaction mixture. After adding a saturated aqueous solution of NH 4 Cl (50 mL), EtOAc (EtOAc) (5-Cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-((2-carbonyl-1) 3-oxoheptyl-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide (65 mg, 86%).
Step 5: (R)-N-(5-Cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-formyl-6-((2) Synthesis of -carbonyl-1,3-oxoheptyl-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide
[0068]
[0069]
(R)-N-(5-Cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-( (2-carbonyl-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide (65 mg, 0.12 mmol) Dissolved in THF/water (volume ratio: 11/4, 4.5 mL), concentrated HCl (0.45 mL, 5.4 mmol), and allowed to react at room temperature for 2 h. Saturated NaHC03 . 3 solution (50mL), (50mL × 2 ) and extracted with ethyl acetate, the organic phases were combined and washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated by column chromatography to give the title compound (R) -N- ( 5-cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-formyl-6-((2-carbonyl-1,3-oxazepine) 3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1 (2H)-carboxamide (30 mg, 51%).
Novel crystalline salt (such as hydrochloride, sulfate, methane sulfonate, mesylate, besylate, ethanesulfonate, oxalate, maleate, p-toluenesulfonate) forms of FGFR4 inhibitor, particularly N-[5-cyano-4-[[(1R)-2-methoxy-1-methyl-ethyl]amino]-2-pyridyl]-7-formyl-6-[(2-oxo-1,3-oxazepan-3-yl)methyl]-3,4-dihydro-2H-1,8-naphthyridine-1-carboxamide (designated as Forms I- IX), compositions comprising them and their use as an FGFR4 inhibitor for the treatment of cancer such as liver cancer, gastric cancer, prostate cancer, skin cancer, ovarian cancer, lung cancer, breast cancer, colon cancer and glioma or rhabdomyosarcoma are claimed.
Example 1: Preparation of a compound of formula (I)
First step 4-(((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)amino)butane Preparation of 1-propanol
2-(Dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-carbaldehyde (1.0 g, 4.2 mmol), 4-aminobutyl at room temperature l-ol (0.45g, 5.1mmol) was dissolved in DCE (15mL), stirred for 2 hours, followed by addition of NaBH (OAc) . 3 (1.35 g of, 6.4 mmol), stirred at room temperature overnight. The reaction was treated with CH 2 CI 2 was diluted (100 mL), the organic phase was washed with water (10mL) and saturated brine (15mL), and dried over anhydrous sodium sulfate, and concentrated by column chromatography to give compound 4 – (((2- ( Dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)amino)butan-1-ol (0.9 g, 69%) .
The second step is 3-((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)-1,3- Preparation of oxazepine-2 ketone
4-(((2-(Dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)amino) in an ice water bath Butan-1-ol (0.6 g, 1.94 mmol) was dissolved in DCE (15 mL), then bis(trichloromethyl) carbonate (0.22 g, 0.76 mmol) was added and triethylamine (0.78 g, 7.76) was slowly added dropwise. Methyl) and then stirred at room temperature for 3 hours. The reaction temperature was raised to 80 ° C, and the reaction was carried out at 80 ° C for 6 hours. After the reaction was cooled to room temperature, it was diluted with CH 2 Cl 2 (100 mL), and the organic phase was washed sequentially with water (10 mL) and brine (15 mL) Drying with sodium sulfate, concentration and column chromatography to give the compound 3-((2-(dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl) )methyl)-1,3-oxazepin-2-one (0.37 g, 57%).
MS m/z (ESI): 336.2 [M+H] + .
The third step is phenyl 7-(dimethoxymethyl)-6-((2-carbonyl-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1, Preparation of 8-naphthyridin-1(2H)-carboxylate
3-((2-(Dimethoxymethyl)-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)methyl)-1,3-oxan -2-one (670mg, 2mmol), diphenyl carbonate (643mg, 3mmol) mixing in of THF (15 mL), N 2 in an atmosphere, cooled to -78 deg.] C, was added dropwise LiHMDS in THF (4mL, 4mmol) was Naturally, it was allowed to react to room temperature overnight. After adding saturated aqueous NH 4 Cl (100 mL), ethyl acetate (100 mL×2), EtOAc. Methyl)-6-((3-carbonylmorpholino)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxylate (432 mg, 47%) .
The fourth step: (R)-N-(5-cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-(dimethoxymethyl) -6-((2-carbonyl-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide synthesis
(R)-6-Amino-4-((1-methoxypropan-2-yl)amino) nicotinenitrile (30 mg, 0.14 mmol), phenyl 7-(dimethoxymethyl)-6- ( (2-carbonyl-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxylate (60 mg, 0.13 Methyl acetate was dissolved in THF (5 mL), cooled to -78 ° C under N 2atmosphere, and a solution of THF (0.3 mL, 0.3 mmol) of LiHMDS was added dropwise to the reaction mixture. After adding a saturated aqueous solution of NH 4 Cl (50 mL), EtOAc (EtOAc) (5-Cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-((2-carbonyl-1) 3-oxoheptyl-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide (65 mg, 86%).
Step 5: (R)-N-(5-Cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-formyl-6-((2) Synthesis of -carbonyl-1,3-oxoheptyl-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide
(R)-N-(5-Cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-(dimethoxymethyl)-6-( (2-carbonyl-1,3-oxazepine-3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1(2H)-carboxamide (65 mg, 0.12 mmol) Dissolved in THF/water (volume ratio: 11/4, 4.5 mL), concentrated HCl (0.45 mL, 5.4 mmol), and allowed to react at room temperature for 2 h. Saturated NaHC03 . 3 solution (50mL), (50mL × 2 ) and extracted with ethyl acetate, the organic phases were combined and washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated by column chromatography to give the title compound (R) -N- ( 5-cyano-4-((1-methoxypropan-2-yl)amino)pyridin-2-yl)-7-formyl-6-((2-carbonyl-1,3-oxazepine) 3-yl)methyl)-3,4-dihydro-1,8-naphthyridin-1 (2H)-carboxamide (30 mg, 51%).









































 The Bangalore-based Aurigene Discovery Technologies Limited, an independent subsidiary of Dr Reddy’s, is now competing in a mature contract research organization space in the country. Established in 2003, Aurigene Discovery Technologies is a partnership focused collaborative discovery organisation. CSN Murthy is chief executive officer of the company. In an interaction with Nandita Vijay, Murthy cut a clear picture of the contract research scene. Excerpts:
The Bangalore-based Aurigene Discovery Technologies Limited, an independent subsidiary of Dr Reddy’s, is now competing in a mature contract research organization space in the country. Established in 2003, Aurigene Discovery Technologies is a partnership focused collaborative discovery organisation. CSN Murthy is chief executive officer of the company. In an interaction with Nandita Vijay, Murthy cut a clear picture of the contract research scene. Excerpts:



























 Integrifolin
Integrifolin






 Journal title :
Journal title : 


















 Chidamide (Epidaza) is an
Chidamide (Epidaza) is an 











 To a suspension of 0.29 g (1.78 mmol) of N, N’-carbonyldiimidazole in tetrahydrofunan (15 ml) is added 0.50 g (1.78 mmol) of 4-[N-(Pyridn-3-ylacryloyl)aminomethyl]benzoic acid, followed by stirring at 45°C for 1 hour. After cooling, the reaction mixture is added to a separately prepared tetrahydrofunan (10 ml) solution including 0.28 g (2.22 mmol) of 4-fluoro-1,2-phenylenediamine and 0.20 g (1.78 mmol) of trifluoroacetic acid at room temperature. After reaction at room temperature for 24 hours, the deposited white solid is collected by filtration, washed with tetrahydrofunan, and then dried to give the title compound (0.40 g, 57%). 1H NMR (300 MHz, DMSO-d6): δppm: 4.49 (2H, d), 4.84 (2H, br.s), 6.60 (IH, t), 6.80 (2H, m), 6.96 (IH, t), 7.18 (IH, d), 7.42 (2H, d), 7.52 (IH, d), 7.95 (2H, d), 8.02 (IH, d), 8.56 (IH, d), 8.72 (IH, br. t), 8.78 (IH, s), 9.60 (IH, br.s). IR (KBr) cm“1: 3310, 1655, 1631, 1524, 1305, 750. HRMS calcd for C22Hι9N4O2F: 390.4170. Found: 390.4172. MA calcd for C22Hι9N4O2F: C, 67.68%; H, 4.40%; N, 14.35. Found: C, 67.52%; H, 4.38%; N, 14.42%.
To a suspension of 0.29 g (1.78 mmol) of N, N’-carbonyldiimidazole in tetrahydrofunan (15 ml) is added 0.50 g (1.78 mmol) of 4-[N-(Pyridn-3-ylacryloyl)aminomethyl]benzoic acid, followed by stirring at 45°C for 1 hour. After cooling, the reaction mixture is added to a separately prepared tetrahydrofunan (10 ml) solution including 0.28 g (2.22 mmol) of 4-fluoro-1,2-phenylenediamine and 0.20 g (1.78 mmol) of trifluoroacetic acid at room temperature. After reaction at room temperature for 24 hours, the deposited white solid is collected by filtration, washed with tetrahydrofunan, and then dried to give the title compound (0.40 g, 57%). 1H NMR (300 MHz, DMSO-d6): δppm: 4.49 (2H, d), 4.84 (2H, br.s), 6.60 (IH, t), 6.80 (2H, m), 6.96 (IH, t), 7.18 (IH, d), 7.42 (2H, d), 7.52 (IH, d), 7.95 (2H, d), 8.02 (IH, d), 8.56 (IH, d), 8.72 (IH, br. t), 8.78 (IH, s), 9.60 (IH, br.s). IR (KBr) cm“1: 3310, 1655, 1631, 1524, 1305, 750. HRMS calcd for C22Hι9N4O2F: 390.4170. Found: 390.4172. MA calcd for C22Hι9N4O2F: C, 67.68%; H, 4.40%; N, 14.35. Found: C, 67.52%; H, 4.38%; N, 14.42%.




















































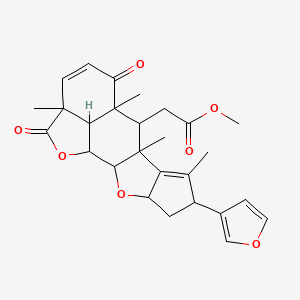






 Dr Gautam Sethi
Dr Gautam Sethi











































 2100300-72-7 CAS
2100300-72-7 CAS



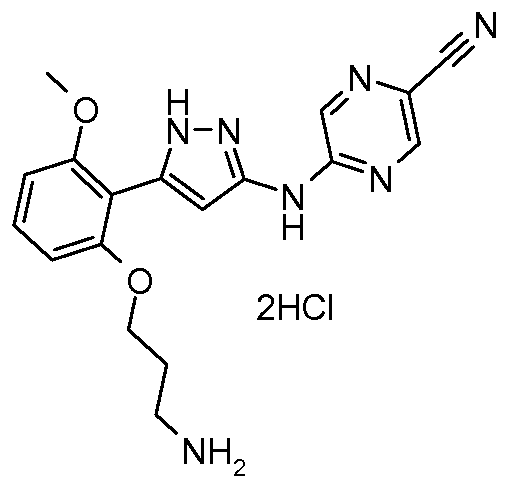




















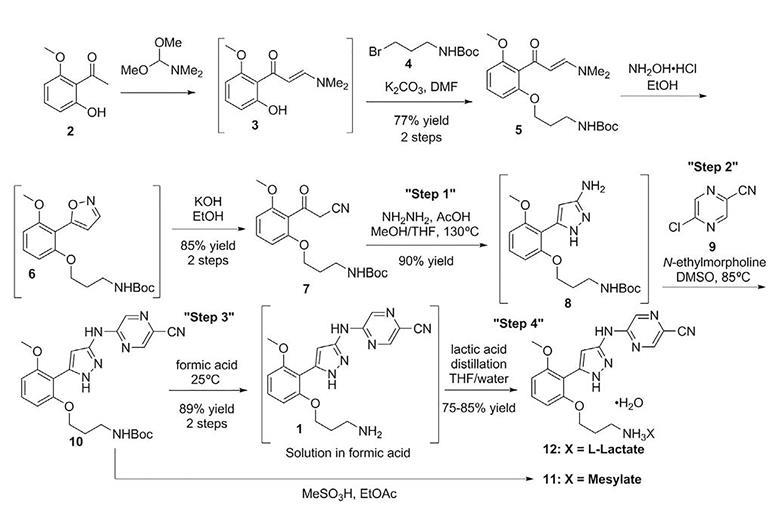





























 and concentrated. Column chromatography of the resulting residue using hexane/ethyl acetate (0- 30%) afforded tert-butyl N- [2- [ [2-chloro-5 -(3 ,3 -diethoxyprop- 1 -ynyl)pyrimidin-4-yl]amino] ethyl] carbamate. 1HNMR (d6-DMSO) δ ppm 8.18 (s, 1H), 7.63 (brs, 1H), 7.40 (brs, 1H), 5.55 (s, 1H), 3.70 (m, 2H), 3.60 (m, 2H), 3.42 (m, 2H), 3.15 (m, 2H), 1.19 – 1.16 (m, 15H). LCMS (ESI) 399 (M + H).
and concentrated. Column chromatography of the resulting residue using hexane/ethyl acetate (0- 30%) afforded tert-butyl N- [2- [ [2-chloro-5 -(3 ,3 -diethoxyprop- 1 -ynyl)pyrimidin-4-yl]amino] ethyl] carbamate. 1HNMR (d6-DMSO) δ ppm 8.18 (s, 1H), 7.63 (brs, 1H), 7.40 (brs, 1H), 5.55 (s, 1H), 3.70 (m, 2H), 3.60 (m, 2H), 3.42 (m, 2H), 3.15 (m, 2H), 1.19 – 1.16 (m, 15H). LCMS (ESI) 399 (M + H).















































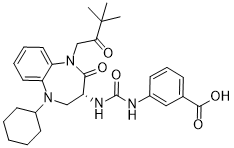
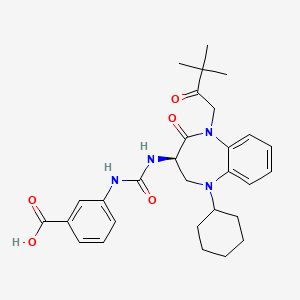



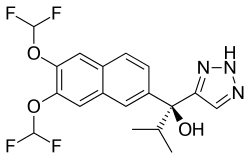

![(E)-3-[6-[2-(6-Methylpyridin-2-yl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl]-[1,2,4]triazolo[1,5-a]pyridin-5-yl]prop-2-enamide.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=132246793&t=l)







