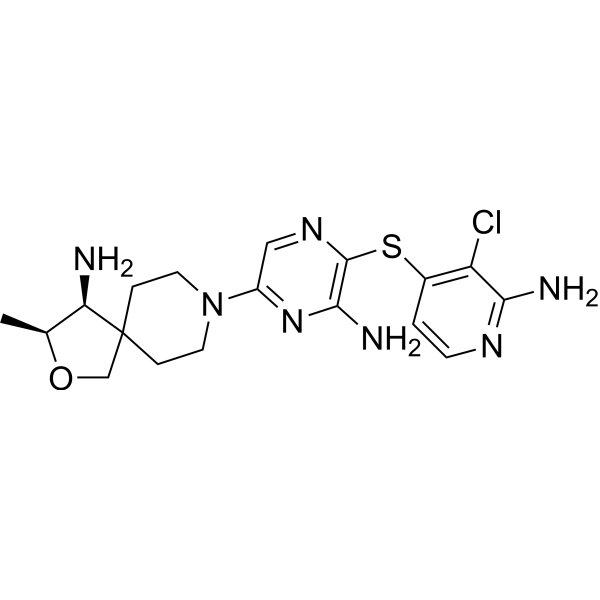
No development reported B-cell lymphoma; Lymphoid leukaemia
26 Mar 2019 National Cancer Institute plans a phase II trial for Cholangiocarcinoma (Combination therapy, Second-line therapy or greater) and Solid tumours (Combination therapy, Second-line therapy or greater) in March 2019 (NCT03878095)
18 Mar 2019 Royal Marsden NHS Foundation Trust and AstraZeneca re-initiate the phase I PATRIOT trial in Solid tumours (Second-line therapy or greater) in United Kingdom (NCT02223923)
25 Dec 2018 University of Michigan Cancer Center plans the phase II TRAP trial for Prostate cancer (Combination therapy; Metastatic disease; Second-line therapy or greater) in February 2019 (NCT03787680)
Inhibits ATR kinase.
Ceralasertib, also known as AZD6738, is an orally available morpholino-pyrimidine-based inhibitor of ataxia telangiectasia and rad3 related (ATR) kinase, with potential antineoplastic activity. Upon oral administration, ATR kinase inhibitor Ceralasertib selectively inhibits ATR activity by blocking the downstream phosphorylation of the serine/threonine protein kinase CHK1. This prevents ATR-mediated signaling, and results in the inhibition of DNA damage checkpoint activation, disruption of DNA damage repair, and the induction of tumor cell apoptosis.
ATR (also known as FRAP-Related Protein 1; FRP1; MEC1; SCKL; SECKL1) protein kinase is a member of the PI3 -Kinase like kinase (PIKK) family of proteins that are involved in repair and maintenance of the genome and its stability (reviewed in Cimprich K.A. and Cortez D. 2008, Nature Rev. Mol. Cell Biol. 9:616-627). These proteins co-ordinate response to DNA damage, stress and cell-cycle perturbation. Indeed ATM and ATR, two members of the family of proteins, share a number of downstream substrates that are themselves recognised components of the cell cycle and DNA-repair machinery e.g. Chkl, BRCAl, p53 (Lakin ND et al,1999, Oncogene; Tibbets RS et al, 2000, Genes & Dev.). Whilst the substrates of ATM and ATR are to an extent shared, the trigger to activate the signalling cascade is not shared and ATR primarily responds to stalled replication forks (Nyberg K.A. et al., 2002, Ann. Rev.
Genet. 36:617-656; Shechter D. et al. 2004, DNA Repair 3:901-908) and bulky DNA damage lesions such as those formed by ultraviolet (UV) radiation (Wright J. A. et al, 1998, Proc. Natl. Acad. Sci. USA, 23:7445-7450) or the UV mimetic agent, 4-nitroquinoline-1-oxi-e, 4NQO (Ikenaga M. et al. 1975, Basic Life Sci. 5b, 763-771). However, double strand breaks (DSB) detected by ATM can be processed into single strand breaks (SSB) recruiting ATR; similarly SSB, detected by ATR can generate DSB, activating ATM. There is therefore a significant interplay between ATM and ATR.
Mutations of the ATR gene that result in complete loss of expression of the ATR protein are rare and in general are not viable. Viability may only result under heterozygous or hypomorphic conditions. The only clear link between ATR gene mutations and disease exists in a few patients with Seckel syndrome which is characterized by growth retardation and microcephaly (O’Driscoll M et al, 2003 Nature Genet. Vol3, 497-501). Cells from patients with hypomorphic germline mutations of ATR (seckel syndrome) present a greater susceptibility to chromosome breakage at fragile sites in presence of replication stress compared to wild type cells (Casper 2004). Disruption of the ATR pathway leads to genomic instability. Patients with Seckel syndrome also present an increased incidence of cancer,suggestive of the role of ATR in this disease in the maintenance of genome stability .
Moreover, duplication of the ATR gene has been described as a risk factor in rhabdomyosarcomas (Smith L et al, 1998, Nature Genetics 19, 39-46). Oncogene-driven tumorigenesis may be associated with ATM loss-of- function and therefore increased reliance on ATR signalling (Gilad 2010). Evidence of replication stress has also been reported in several tumour types such as colon and ovarian cancer, and more recently in glioblastoma, bladder, prostate and breast (Gorgoulis et al, 2005; Bartkova et al. 2005a; Fan et al., 2006; Tort et al, 2006; Nuciforo et al, 2007; Bartkova et al., 2007a). Loss of Gl checkpoint is also frequently observed during tumourigenesis. Tumour cells that are deficient in Gl checkpoint controls, in particular p53 deficiency, are susceptible to inhibition of ATR activity and present with premature chromatin condensation (PCC) and cell death (Ngheim et al, PNAS, 98, 9092-9097).
ATR is essential to the viability of replicating cells and is activated during S-phase to regulate firing of replication origins and to repair damaged replication forks (Shechter D et al, 2004, Nature cell Biology Vol 6 (7) 648-655). Damage to replication forks may arise due to exposure of cells to clinically relevant cytotoxic agents such as hydroxyurea (HU) and platinums (O’Connell and Cimprich 2005; 118, 1-6). ATR is activated by most cancer chemotherapies (Wilsker D et al, 2007, Mol. Cancer Ther. 6(4) 1406-1413). Biological assessment of the ability of ATR inhibitors to sensitise to a wide range of chemotherapies have been evaluated. Sensitisation of tumour cells to chemotherapeutic agents in cell growth assays has been noted and used to assess how well weak ATR inhibitors (such as Caffeine) will sensitise tumour cell lines to cytotoxic agents. (Wilsker D .et al, 2007, Mol Cancer Ther. 6 (4)1406-1413; Sarkaria J.N. et al, 1999, Cancer Res. 59, 4375-4382). Moreover, a reduction of ATR activity by siRNA or ATR knock-in using a dominant negative form of ATR in cancer cells has resulted in the sensitisation of tumour cells to the effects of a number of therapeutic or experimental agents such as antimetabolites (5-FU, Gemcitabine, Hydroxyurea, Metotrexate, Tomudex), alkylating agents (Cisplatin, Mitomycin C, Cyclophosphamide, MMS) or double-strand break inducers (Doxorubicin, Ionizing radiation) (Cortez D. et al. 2001, Science, 294:1713-1716; Collis S.J. et al, 2003, Cancer Res. 63:1550-1554; Cliby W.A. et al, 1998, EMBO J. 2:159-169) suggesting that the combination of ATR inhibitors with some cytotoxic agents might be therapeutically beneficial.
An additional phenotypic assay has been described to define the activity of specific ATR inhibitory compounds is the cell cycle profile (PJ Hurley, D Wilsker and F Bunz, Oncogene, 2007, 26, 2535-2542). Cells deficient in ATR have been shown to have defective cell cycle regulation and distinct characteristic profiles, particularly following a cytotoxic cellular insult. Furthermore, there are proposed to be differential responses between tumour and normal tissues in response to modulation of the ATR axis and this provides further potential for therapeutic intervention by ATR inhibitor molecules (Rodnguez-Bravo V et al, Cancer Res., 2007, 67, 11648-11656).
Another compelling utility of ATR-specific phenotypes is aligned with the concept of synthetic lethality and the observation that tumour cells that are deficient in G1 checkpoint controls, in particular p53 deficiency, are susceptible to inhibition of ATR activity resulting in premature chromatin condensation (PCC) and cell death (Ngheim et al, PNAS, 98, 9092-9097). In this situation, S-phase replication of DNA occurs but is not completed prior to M-phase initiation due to failure in the intervening checkpoints resulting in cell death from a lack of ATR signalling. The G2/M checkpoint is a key regulatory control involving ATR (Brown E. J. and Baltimore D., 2003, Genes Dev. 17, 615-628) and it is the compromise of this checkpoint and the prevention of ATR signalling to its downstream partners which results in PCC. Consequently, the genome of the daughter cells is compromised and viability of the cells is lost (Ngheim et al, PNAS, 98, 9092-9097).
It has thus been proposed that inhibition of ATR may prove to be an efficacious approach to future cancer therapy (Collins I. and Garret M.D., 2005, Curr. Opin. Pharmacol., 5:366-373; Kaelin W.G. 2005, Nature Rev. Cancer, 5:689-698) in the appropriate genetic context such as tumours with defects in ATM function or other S-phase checkpoints. Until recently, There is currently no clinical precedent for agents targeting ATR, although agents targeting the downstream signalling axis i.e. Chk1 are currently undergoing clinical evaluation (reviewed in Janetka J.W. et al. Curr Opin Drug Discov Devel, 2007, 10:473-486). However, inhibitors targeting ATR kinase have recently been described (Reaper 2011, Charrier 2011).
In summary ATR inhibitors have the potential to sensitise tumour cells to ionising radiation or DNA-damage inducing chemotherapeutic agents, have the potential to induce selective tumour cell killing as well as to induce synthetic lethality in subsets of tumour cells with defects in DNA damage response.
PAPER
Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent
The kinase ataxia telangiectasia mutated and rad3 related (ATR) is a key regulator of the DNA-damage response and the apical kinase which orchestrates the cellular processes that repair stalled replication forks (replication stress) and associated DNA double-strand breaks. Inhibition of repair pathways mediated by ATR in a context where alternative pathways are less active is expected to aid clinical response by increasing replication stress. Here we describe the development of the clinical candidate 2(AZD6738), a potent and selective sulfoximine morpholinopyrimidine ATR inhibitor with excellent preclinical physicochemical and pharmacokinetic (PK) characteristics. Compound 2 was developed improving aqueous solubility and eliminating CYP3A4 time-dependent inhibition starting from the earlier described inhibitor 1 (AZ20). The clinical candidate 2 has favorable human PK suitable for once or twice daily dosing and achieves biologically effective exposure at moderate doses. Compound 2 is currently being tested in multiple phase I/II trials as an anticancer agent.
(R)-3-Methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (98 mg, 0.18 mmol) was dissolved in MeOH (10 ml) and DCM (10 ml) and heated to 50 °C. Sodium hydroxide, 2M aqueous solution (0.159 ml, 0.32 mmol) was then added and heating continued for 5 hours. The reaction mixture was evaporated and the residue dissolved in DME: water :MeCN 2: 1 : 1 (4 ml) and then purified by preparative HPLC using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents. Fractions containing the desired compound were evaporated and the residue trituated with Et2O
The (R)-3-methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine, used as starting material, can be prepared as follows:
a) (R)-3-methylmorpholine (7.18 g, 71.01 mmol) and triethylamine (12.87 ml, 92.31 mmol) were added to methyl 2,4-dichloropyrimidine-6-carboxylate (14.70 g, 71.01 mmol) in DCM (100 ml). The resulting mixture was stirred at RT for 18 hours. Water (100 ml) was added, the layers separated and extracted with DCM (3 × 75 ml). The combined organics were
dried over MgSO4, concentrated in vacuo and the residue triturated with Et2O to yield (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (14.77 g, 77%); 1H NMR (400 MHz, CDCl3) 1.35 (3H, d), 3.34 (1H, td), 3.55 (1H, td), 3.70 (1H, dd), 3.81 (1H, d), 3.97 (3H, s), 4.03 (1H, dd), 4.12 (1H, br s), 4.37 (1H, br s), 7.15 (1H, s); m/z: (ESI+) MH+, 272.43. The liquors were concentrated onto silica and purified by chromatography on silica eluting with a gradient of 20 to 40% EtOAc in isohexane. Fractions containing product were combined and evaporated to afford (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (1.659 g, 9%); 1H NMR (400 MHz, CDCl3) 1.35 (3H, d), 3.33 (1H, td), 3.55 (1H, td), 3.69 (1H, dd), 3.80 (1H, d), 3.97 (3H, s), 4.03 (1H, dd), 4.12 (1H, br s), 4.36 (1H, br s), 7.15 (1H, s); m/z: (ESI+) MH+, 272.43.
b) Lithium borohydride, 2M in THF (18 ml, 36.00 mmol) was added dropwise to (R)-methyl 2-chloro-6-(3-methylmorpholino)pyrimidine-4-carboxylate (16.28 g, 59.92 mmol) in THF (200 ml) at 0°C over a period of 20 minutes under nitrogen. The resulting solution was stirred at 0 °C for 30 minutes and then allowed to warm to RT and stirred for a further 18 hours. Water (200 ml) was added and the THF evaporated. The aqueous layer was extracted with EtOAc (2 × 100 ml) and the organic phases combined, dried over MgSO4 and then evaporated to afford (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (14.54 g, 100%) which was used in the next step without purification; 1HNMR (400 MHz, CDCl3) 1.32 (3H, d), 2.65 (1H, br s), 3.25 – 3.32 (1H, m), 3.51 – 3.57 (1H, m), 3.67 – 3.70 (1H, m), 3.78 (1H, d), 3.98 – 4.09 (2H, m), 4.32 (1H, br s), 4.59 (2H, s), 6.44 (1H, s); m/z: (ESI+) MH+, 244.40.
c) Methanesulfonyl chloride (4.62 ml, 59.67 mmol) was added dropwise to (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (14.54 g, 59.67 mmol) and triethylamine (8.32 ml, 59.67 mmol) in DCM (250 ml) at 25 °C over a period of 5 minutes. The resulting solution was stirred at 25 °C for 90 minutes. The reaction mixture was quenched with water (100 ml) and extracted with DCM (2 × 100 ml). The organic phases were combined, dried over MgSO4, filtered and evaporated to afford (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (20.14 g, 105%) which was used in the next step without further purification; 1H NMR (400 MHz, CDCl3) 1.33 (3H, d), 3.13 (3H, s), 3.27 – 3.34 (1H, m), 3.51 -3.57 (1H, m), 3.66 – 3.70 (1H, m), 3.79 (1H, d), 3.99 – 4.03 (2H, m), 4.34 (1H, br s), 5.09 (2H, d) , 6.52 (1H, s); m/z: (ESI+) MH+, 322.83.
Alternatively, this step can be carried out as follows:
In a 3 L fixed reaction vessel with a Huber 360 heater / chiller attached, under a nitrogen atmosphere, triethylamine (0.120 L, 858.88 mmol) was added in one go to a stirred solution of (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methanol (161 g, 660.68 mmol) in DCM (7.5vol) (1.2 L) at 20°C (3°C exotherm seen). The mixture was cooled to 5°C and then methanesulfonyl chloride (0.062 L, 792.81 mmol) was added dropwise over 15 minutes, not allowing the internal temperature to exceed 15°C. The reaction mixture was stirred at 15°C for 2 hours and then held (not stirring) overnight at RT under a nitrogen atmosphere. Water (1.6 L, 10 vol) was added and the aqueous layer was separated and then extracted with DCM (2 × 1.6 L, 2 × 10 vol). The organics were combined, washed with 50% brine / water (1.6 L, 10 vol), dried over magnesium sulphate, filtered and then evaporated to afford a mixture of
approximately two thirds (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate and one third (R)-4-(2-chloro-6-(chloromethyl)pyrimidin-4-yl)-3-methylmorpholine (216 g) which was used in the next step without further purification, d) Lithium iodide (17.57 g, 131.27 mmol) was added to (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (19.2 g, 59.67 mmol) in dioxane (300 ml) and heated to 100 °C for 2 hours under nitrogen. The reaction mixture was quenched with water (200 ml) and extracted with EtOAc (3 × 200 ml). The organic layers were combined and washed with 2M sodium bisulfite solution (400 ml), water (400 ml), brine (400 ml) dried over MgSO4 and then evaporated. The residue was triturated with Et2O to afford (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (13.89 g, 66%); 1H NMR (400 MHz, CDCl3) 1.32 (3H, d), 3.28 (1H, td), 3.54 (1H, td), 3.69 (1H, dd), 3.78 (1H, d), 3.98 -4.02 (2H, m), 4.21 (2H, s), 4.29 (1H, br s), 6.41 (1H, s); m/z: (ESI+) MH+ 354.31.
The mother liquors were concentrated down and triturated with Et2O to afford a further crop of (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (2.46 g, 12%); 1HNMR (400 MHz, CDCI3) 1.32 (3H, d), 3.28 (1H, td), 3.54 (1H, td), 3.69 (1H, dd), 3.78 (1H, d), 3.98 – 4.02 (2H, m), 4.21 (2H, s), 4.30 (1H, s), 6.41 (1H, s); m/z: (ESI+) MH+, 354.31.
Alternatively, this step can be carried out as follows:
(R)-(2-Chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate (80 g, 248.62 mmol) and lithium iodide (83 g, 621.54 mmol) were dissolved in dioxane (300 ml) and then heated at 107 °C for 1 hour. The reaction mixture was quenched with water (250 ml), extracted with EtOAc (3 × 250 ml), the organic layer was dried over MgSO4, filtered and evaporated. The residue was dissolved in DCM and Et2O was added, the mixture was passed through silica (4 inches) and eluted with Et2O. Fractions containing product were evaporated and the residue was then triturated with Et2O to give a solid which was collected by filtration and dried under vacuum to afford (R)-4-(2-chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (75 g, 86%) ; m/z: (ESI+) MH+, 354.27.
e) (R)-4-(2-Chloro-6-(iodomethyl)pyrimidin-4-yl)-3-methylmorpholine (17.0 g, 48.08 mmol) was dissolved in DMF (150 ml), to this was added sodium methanethiolate (3.37 g, 48.08 mmol) and the reaction was stirred for 1 hour at 25 °C. The reaction mixture was quenched with water (50 ml) and then extracted with Et2O (3 × 50 ml). The organic layer was dried over MgSO4, filtered and then evaporated. The residue was purified by flash
chromatography on silica, eluting with a gradient of 50 to 100% EtOAc in iso-hexane. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (12.63 g, 96%); m/z: (ES+) MH+, 274.35.
Alternatively, (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine, may be prepared as follows:
In a 3 L fixed vessel, sodium thiomethoxide (21% in water) (216 g, 646.69 mmol) was added dropwise over 5 minutes to a stirred solution of a mixture of approximately two thirds (R)-(2-chloro-6-(3-methylmorpholino)pyrimidin-4-yl)methyl methanesulfonate and one third (R)-4-(2-chloro-6-(chloromethyl)pyrimidin-4-yl)-3-methylmorpholine (130.2 g, 431 mmol) and sodium iodide (1.762 ml, 43.11 mmol) in MeCN (1 L) at RT (temperature dropped from 20 °C to 18 °C over the addition and then in the next 5 minutes rose to 30 °C). The reaction mixture was stirred for 16 hours and then diluted with EtOAc (2 L), and washed sequentially with water (750 ml) and saturated brine (1 L). The organic layer was dried over MgSO4, filtered and then evaporated to afford (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (108 g, 91%); 1H NMR (400 MHz, DMSO- d6) 1.20 (3H, d), 2.07 (3H, s), 3.11 – 3.26 (1H, m), 3.44 (1H, td), 3.53 (2H, s), 3.59 (1H, dd), 3.71 (1H, d), 3.92 (1H, dd), 3.92 – 4.04 (1H, br s), 4.33 (1H, s), 6.77 (1H, s); m/z: (ES+) MH+, 274.36.
f) (R)-4-(2-Chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (12.63 g, 46.13 mmol) was dissolved in DCM (100 ml), to this was added mCPBA (7.96 g, 46.13 mmol) in one portion and the reaction mixture was stirred for 10 minutes at 25 °C. An additional portion of mCPBA (0.180 g) was added. The reaction mixture was quenched with saturated Na2CO3 solution (50 ml) and extracted with DCM (3 × 50 ml). The organic layer was dried over MgSO4, filtered and then evaporated. The residue was dissolved in DCM (80 ml) in a 150
ml conical flask which was placed into a beaker containing Et2O (200 ml) and the system covered with laboratory film and then left for 3 days. The obtained crystals were filtered, crushed and sonicated with Et2O. The crystallisation procedure was repeated to afford (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine as white needles (3.87 g, 29%); 1HNMR (400 MHz, CDCl3) 1.33 (3H, d), 2.62 (3H, s), 3.30 (1H, td), 3.53 (1H, td), 3.68 (1H, dd), 3.76 (2H, dd), 3.95 (1H, d), 4.00 (1H, dd), 4.02 (1H, s), 4.32 (1H, s), 6.42 (1H, s).
The remaining liquour from the first vapour diffusion was purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine as an orange gum (5.70 g, 43%); 1 HNMR (400 MHz, CDCl3) 1.33 (3H, d), 2.62 (3H, d), 3.29 (1H, td), 3.54 (1H, td), 3.68 (1H, dd), 3.73 – 3.82 (2H, m), 3.94 (1H, dd), 4.00 (2H, dd), 4.33 (1H, s), 6.42 (1H, s).
Alternatively, this step can be carried out as follows:
Sodium meta-periodate (64.7 g, 302.69 mmol) was added in one portion to (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (82.87 g, 302.69 mmol) in water (500 ml), EtOAc (1000 ml) and MeOH (500 ml). The resulting solution was stirred at 20 °C for 16 hours. Sodium metabisulfite (50 g) was added and the mixture stirred for 30 minutes. The reaction mixture was filtered and then partially evaporated to remove the MeOH. The organic layer was separated, dried over MgSO4, filtered and then evaporated. The aqueous layer was washed with DCM (3 x 500 ml). The organic layers were combined, dried over MgSO4, filtered and then evaporated. The residues were combined and dissolved in DCM (400 ml) and purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Fractions containing product were evaporated and the residue was dissolved in DCM (400 ml) and then divided into four 450 ml bottles. An aluminium foil cap was placed over the top of each bottle and a few holes made in each cap. The bottles were placed in pairs in a large dish containing Et2O (1000 ml), and then covered and sealed with a second glass dish and left for 11 days. The resultant white needles were collected by filtration and dried under vacuum. The crystals were dissolved in DCM (200 ml) and placed into a 450 ml bottle. An aluminium foil cap was placed over the top of the bottle and a few holes made in the cap. The bottle was placed in a large dish containing Et2O (1500 ml) and then covered and sealed with a second glass dish and left for 6 days. The resultant crystals were collected by filtration and dried under vacuum to afford (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (16.53 g, 19%); 1H NMR (400 MHz, CDCl3) 1.33 (3H, d), 2.61 (3H, s),
The filtrate from the first vapour diffusion was concentrated in vacuo to afford an approximate
5:2 mixture of (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine and (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (54.7 g, 62%).
Alternatively, this step can be carried out as follows:
Sodium meta-periodate (2.87 g, 13.44 mmol) was added in one portion to (R)-4-(2-chloro-6-(methylthiomethyl)pyrimidin-4-yl)-3-methylmorpholine (3.68 g, 13.44 mmol) in water (10.00 ml), EtOAc (20 ml) and MeOH (10.00 ml). The resulting solution was stirred at 20 °C for 16 hours. The reaction mixture was diluted with DCM (60 ml) and then filtered. The DCM layer was separated and the aqueous layer washed with DCM (3 × 40 ml). The organics were combined, dried over MgSO4, filtered and then evaporated. The residue was purified by flash chromatography on silica, eluting with a gradient of 0 to 7% MeOH in DCM. Pure fractions were evaporated to afford (R)-4-(2-chloro-6-(methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (2.72 g, 70%); 1H NMR (400 MHz, DMSO-d6) 1.22 (3H, d), 2.64 (3H, d), 3.14 – 3.26 (1H, m), 3.45 (1H, td), 3.59 (1H, dd), 3.73 (1H, d), 3.88 – 3.96 (2H, m), 4.00 (1H, d), 4.07 (1H, dt), 4.33 (1H, s), 6.81 (1H, s); m/z: (ESI+) MH+, 290.43.
The (3R)-4-(2-chloro-6-(methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (2.7 g, 9.32 mmol) was purified by preparative chiral chromatography on a Merck 100 mm 20 μm Chiralpak AD column, eluting isocratically with a 50:50:0.1 mixture of iso-Hexane:EtOH:TEA as eluent. The fractions containing product were evaporated to afford (R)-4-(2-chloro-6-((S)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (1.38 g, 51%) as the first eluting compound; 1HNMR (400 MHz, CDCl3) 1.29 (3H, dd), 2.56 (3H, s), 3.15 – 3.33 (1H, m), 3.46 (1H, tt), 3.55 – 3.83 (3H, m), 3.85 – 4.06 (3H, m), 4.31 (1H, s), 6.37 (1H, s). Chiral HPLC: (HP1100 System 6, 20μm Chiralpak AD (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/TEA 50/50/0.1) Rf, 7.197 >99%.
and (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (1.27 g, 47 %) as the second eluting compound; 1H NMR (400 MHz, CDCl3) 1.28 (3H, d), 2.58 (3H, s),
3.26 (1H, td), 3.48 (1H, td), 3.62 (1H, dt), 3.77 (2H, dd), 3.88 – 4.13 (3H, m), 4.28 (1H, s), 6.37 (1H, s). Chiral HPLC: (HP1100 System 6, 20μm Chiralpak AD (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/TEA 50/50/0.1) Rf, 16.897 >99%.
g) Iodobenzene diacetate (18.98 g, 58.94 mmol) was added to (R)-4-(2-chloro-6-((R)-methylsulfinylmethyl)pyrimidin-4-yl)-3-methylmorpholine (17.08 g, 58.94 mmol), 2,2,2-trifluoroacetamide (13.33 g, 117.88 mmol), magnesium oxide (9.50 g, 235.76 mmol) and rhodium(II) acetate dimer (0.651 g, 1.47 mmol) in DCM (589 ml) under air. The resulting suspension was stirred at 20 °C for 24 hours. Further 2,2,2-trifluoroacetamide (13.33 g, 117.88 mmol), magnesium oxide (9.50 g, 235.76 mmol), iodobenzene diacetate (18.98 g, 58.94 mmol) and rhodium(II) acetate dimer (0.651 g, 1.47 mmol) were added and the suspension was stirred at 20 °C for 3 days. The reaction mixture was filtered and then silica gel (100 g) added to the filtrate and the solvent removed in vacuo. The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 20 to 50% EtOAc in isohexane. Pure fractions were evaporated to afford N-[({2-chloro-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}methyl)(methyl)oxido-λ6-(R)-sulfanylidene]-2,2,2-trifluoroacetamide (19.39 g, 82%); 1H NMR (400 MHz, DMSO-d6) 1.22 (3H, d), 3.17 – 3.27 (1H, m), 3.44 (1H, td), 3.59 (1H, dd), 3.62 (3H, s), 3.74 (1H, d), 3.95 (1H, dd), 4.04 (1H, br s), 4.28 (1H, s), 5.08 (2H, q), 6.96 (1H, s); m/z: (ESI+) MH+, 401.12 and 403.13.
h) Dichlorobis(triphenylphosphine)palladium(II) (8.10 mg, 0.01 mmol) was added in one portion to N-[({2-chloro-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}methyl)(methyl)oxido-λ6-(R)-sulfanylidene]-2,2,2-trifluoroacetamide (185 mg, 0.46 mmol), 2M aqueous Na2CO3 solution (0.277 ml, 0.55 mmol) and 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (193 mg, 0.48 mmol) in DME:water 4: 1 (5 ml) at RT. The reaction mixture was stirred at 90 °C for 1 hour, filtered and then purified by preparative HPLC using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents. Fractions containing the desired compound were evaporated to afford (R)-3-methyl-4-(6-((R)-S-methylsulfonimidoylmethyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (102 mg, 41%); 1HNMR (400 MHz, CDCl3) 1.33 (3H, d), 3.21 – 3.38 (1H, m), 3.42 (3H, d), 3.45 – 3.57 (1H, m), 3.61 – 3.70 (1H, m), 3.78 (1H, d), 4.01 (1H, dd), 3.90 -4.15 (1H, br s), 4.30 (1H, s), 4.64 (1H, dd), 4.84 (1H, dd), 6.49 (1H, d); m/z: (ESI+) MH+, 541.35
The 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine, used as starting material, can be prepared as follows:
a) To a 3L fixed vessel was charged 3-chlorobenzoperoxoic acid (324 g, 1444.67 mmol) portionwise to 1H-pyrrolo[2,3-b]pyridine (150 g, 1244.33 mmol) in DME (750 ml) and heptane (1500 ml) at 20°C over a period of 1 hour under nitrogen. The resulting slurry was stirred at 20 °C for 18 hours. The precipitate was collected by filtration, washed with DME / heptane (1/2 5 vol) (750 ml) and dried under vacuum at 40°C to afford 1H-pyrrolo[2,3-b] pyridine 7-oxide 3-chlorobenzoate (353 g, 97%) as a cream solid, which was used without further purification; 1H NMR (400 MHz, DMSO-d6) 6.59 (1H, d), 7.07 (1H, dd), 7.45 (1H, d), 7.55 (1H, t), 7.65 (1H, dd), 7.70 (1H, ddd), 7.87 – 7.93 (2H, m), 8.13 (1H, d), 12.42 (1H, s), 13.32 (1H, s).
b) A 2M solution of potassium carbonate (910 ml, 1819.39 mmol) was added dropwise to a stirred slurry of 1H-pyrrolo[2,3-b]pyridine 7-oxide 3-chlorobenzoate (352.6 g, 1212.93 mmol) in water (4.2 vol) (1481 ml) at 20°C, over a period of 1 hour adjusting the pH to 10. To the resulting slurry was charged water (2 vol) (705 ml) stirred at 20 °C for 1 hour. The slurry was cooled to 0°C for 1 hour and the slurry filtered, the solid was washed with water (3 vol 1050ml) and dried in a vacuum oven at 40°C over P2O5 overnight to afford 1H-pyrrolo[2,3-b] pyridine 7-oxide (118 g, 73%); 1H NMR (400 MHz, DMSO-d6) 6.58 (1H, d), 7.06 (1H, dd), 7.45 (1H, d), 7.64 (1H, d), 8.13 (1H, d), 12.44 (1H, s); m/z: (ES+) (MH+MeCN)+, 176.03. c) To a 3L fixed vessel under an atmosphere of nitrogen was charged methanesulfonic anhydride (363 g, 2042.71 mmol) portionwise to 1H-pyrrolo[2,3-b]pyridine 7-oxide (137 g, 1021.36 mmol), and tetramethylammonium bromide (236 g, 1532.03 mmol) in DMF (10 vol) (1370 ml) cooled to 0°C over a period of 30 minutes under nitrogen. The resulting suspension was stirred at 20 °C for 24 hours. The reaction mixture was quenched with water (20 vol, 2740 ml) and the reaction mixture was adjusted to pH 7 with 50% sodium hydroxide (approx 200 ml). Water (40 vol, 5480 ml) was charged and the mixture cooled to 10°C for 30 minutes. The solid was filtered, washed with water (20 vol, 2740 ml) and the solid disssolved into
DCM/methanol (4: 1, 2000 ml), dried over MgSO4 and evaporated to provide a light brown solid. The solid was taken up in hot methanol (2000 ml) and water added dropwise until the solution went turbid and left overnight. The solid was filtered off and discarded, the solution was evaporated and the solid recrystallised from MeCN (4000 ml). The solid was filtered and washed with MeCN to afford 4-bromo-1H-pyrrolo[2,3-b]pyridine (68.4 g, 34%) as a pink
solid; 1H NMR (400 MHz, OMSO-d6) 6.40 – 6.45 (1H, m), 7.33 (1H, d), 7.57 – 7.63 (1H, m), 8.09 (1H, t), 12.02 (1H, s); m/z: (ES+) MH+, 198.92. The crude mother liquors were purified by Companion RF (reverse phase CI 8, 415g column), using decreasingly polar mixtures of water (containing 1% NH3) and MeCN as eluents (starting at 26% upto 46% MeCN). Fractions containing the desired compound were evaporated to afford 4-bromo-1H-pyrrolo[2,3-b]pyridine (5.4 g, 3%) as a pink solid; 1H NMR (400 MHz, DMSO-d6) 6.43 (1H, dd), 7.33 (1H, d), 7.55 – 7.66 (1H, m), 8.09 (1H, d), 12.03 (1H, s); m/z: (ES+) MH+, 199.22.
d) Sodium hydroxide (31.4 ml, 188.35 mmol) was added to 4-bromo-1H-pyrrolo[2,3-b]pyridine (10.03 g, 50.91 mmol), tosyl chloride (19.41 g, 101.81 mmol) and
tetrabutylammonium hydrogensulfate (0.519 g, 1.53 mmol) in DCM (250 ml) at RT. The resulting mixture was stirred at RT for 1 hour. The reaction was quenched through the addition of saturated aqueous NH4Cl, the organic layer removed and the aqueous layer further extracted with DCM (3 × 25 ml). The combinbed organics were washed with brine (100 ml), dried over Na2SO4 and then concentrated under reduced pressure. The residue was purified by flash chromatography on silica, eluting with a gradient of 0 to 20% EtOAc in isohexane. Pure fractions were evaporated to afford 4-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (14.50 g, 81%); 1H NMR (400 MHz, CDCl3) 2.38 (3H, s), 6.64 (1H, d), 7.28 (2H, d), 7.36 (1H, d), 7.78 (1H, d), 8.06 (2H, d), 8.22 (1H, d); m/z: (ES+) MH+, 353.23.
e) 1,1′-Bis(diphenylphosphino)ferrocenedichloropalladium(II) (3.37 g, 4.13 mmol) was added in one portion to 4-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (14.5 g, 41.28 mmol), bis(pinacolato)diboron (20.97 g, 82.57 mmol) and potassium acetate (12.16 g, 123.85 mmol) in anhydrous DMF (300 ml) at RT. The resulting mixture was stirred under nitrogen at 90 °C for 24 hours. After cooling to RT, 1N aqueous NaOH was added untill the aqueous layer was taken to pH 10. The aqueous layer was washed with DCM (1L), carefully acidified to pH 4 with 1 N aqueous HCl, and then extracted with DCM (3 × 300 ml). The organic layer was concentrated under reduced pressure to afford a dark brown solid. The solid was triturated with diethyl ether, filtered and dried to afford 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (7.058 g, 43%); 1H NMR (400 MHz, CDCl3) 1.36 (12H, s), 2.35 (3H, s), 7.01 (1H, d), 7.22 (2H, d), 7.52 (1H, d), 7.74 (1H, d), 8.03 (2H, m), 8.42 (1H, d); m/z: (ES+) MH+, 399.40. The mother liquors were concentrated in vacuo and the residue triturated in isohexane, filtered and dried to afford a further sample of 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (3.173 g, 19%); 1H NMR (400 MHz,
(3R)-3-Methyl-4-(6-(1-(S-methylsulfonimidoyl)cyclopropyl)-2-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl)morpholine (1.67 g, 2.95 mmol) was dissolved in DME:water 4: 1 (60 ml) and heated to 50 °C. Sodium hydroxide, 2M aqueous solution (2.58 ml, 5.16 mmol) was then added and heating continued for 18 hours. The reaction mixture was acidified with 2M H Cl (~2 ml) to pH5. The reaction mixture was evaporated to dryness and the residue dissolved in EtOAc (250 ml), and washed with water (200 ml). The organic layer was dried over MgSO4, filtered and evaporated onto silica gel (10 g). The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 0 to 7% MeOH in DCM. Pure fractions were evaporated and the residue was purified by preparative chiral chromatography on a Merck 50mm, 20μm ChiralCel OJ column, eluting isocratically with 50% isohexane in EtOH/MeOH (1 : 1) (modified with TEA) as eluent. The fractions containing the desired compound were evaporated to dryness to afford the title compound: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.538g, 44%) as the first eluting compound; 1H NMR (400 MHz,
DMSO-d6) 1.29 (3H, d), 1.51 (3H, m), 1.70 – 1.82 (1H, m), 3.11 (3H, s), 3.28 (1H, m, obscured by water peak), 3.48 – 3.60 (1H, m), 3.68 (1H, dd), 3.75 – 3.87 (2H, m), 4.02 (1H, dd), 4.19 (1H, d), 4.60 (1H, s), 7.01 (1H, s), 7.23 (1H, dd), 7.51 – 7.67 (1H, m), 7.95 (1H, d), 8.34 (1H, d), 11.76 (1H, s); m/z: (ES+) MH+, 413.12. Chiral HPLC: (HP1100 System 4, 5μm Chiralcel OJ-H (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/MeOH/TEA 50/25/25/0.1) Rf, 9.013 >99%. Crystals were grown and isolated by slow evaporation to dryness in air from EtOAc. These crystals were used to obtain the structure shown in Fig 1 by X-Ray diffraction (see below). Example 2.02: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (326 mg, 0.79 mmol) was dissolved in DCM (3 ml). Silica gel (0.5 g) was added and the mixture concentrated in vacuo. The resulting powder was purified by flash chromatography on silica, eluting with a gradient of 0 to 5% MeOH in DCM. Pure fractions were evaporated to dryness and the residue was crystallized from EtOAc/n-heptane to afford 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((R)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (256 mg, 79%) as a white crystalline solid; 1H NMR (400 MHz, DMSO-d6) 1.29 (3H, d), 1.39 – 1.60 (3H, m), 1.71 – 1.81 (1H, m), 3.10 (3H, d), 3.21 – 3.29 (1H, m), 3.52 (1H, td), 3.67 (1H, dd), 3.80 (2H, t), 4.01 (1H, dd), 4.19 (1H, d), 4.59 (1H, s), 7.01 (1H, s), 7.23 (1H, dd), 7.54 – 7.62 (1H, m), 7.95 (1H, d), 8.34 (1H, d), 11.75 (1H, s). DSC (Mettler-Toledo DSC 820, sample run at a heating rate of 10°C per minute from 30°C to 350°C in a pierced aluminium pan) peak, 224.1 FC.
and the title compound: 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.441 g, 36%) as the second eluting compound; 1H NMR (400 MHz, DMSO-d6) 1.28 (3H, d), 1.40 – 1.58 (3H, m), 1.70 – 1.80 (1H, m), 3.10 (3H, d), 3.23 – 3.27 (1H, m), 3.51 (1H, dt), 3.66 (1H, dd), 3.80 (2H, d), 4.01 (1H, dd), 4.21 (1H, d), 4.56 (1H, s), 6.99 (1H, s), 7.22 (1H, dd), 7.54 – 7.61 (1H, m), 7.94 (1H, d), 8.33 (1H, d), 11.75 (1H, s); m/z: (ES+) MH+, 413.12. Chiral HPLC: (HP1100 System 4, 5μm Chiralcel OJ-H (250 mm × 4.6 mm) column eluting with iso-Hexane/EtOH/MeOH/TEA 50/25/25/0.1) Rf, 15.685 >99%. Example 2.01 : 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (66.5 mg) was purified by crystallisation from EtOH/water to afford 4-{4-[(3R)-3-methylmorpholin-4-yl]-6-[1-((S)-S-methylsulfonimidoyl)cyclopropyl]pyrimidin-2-yl}-1H-pyrrolo[2,3-b]pyridine (0.050 g); 1H NMR (400 MHz, CDCl3) 1.40 (3H, d), 1.59 (2H, s), 1.81 (2H, s), 2.41 (1H, s), 3.16 (3H, s), 3.39 (1H, td), 3.59 – 3.67 (1H, m), 3.77 (1H, dd), 3.86 (1H, d), 4.07 (1H, dd), 4.17 (1H, d), 4.54 (1H, s), 6.91 (1H, s), 7.34 (1H, t), 7.43 (1H, t), 8.05 (1H, d), 8.41 (1H, d), 9.14 (1H, s).
Scheme 1. Medicinal Chemistry Route to AZD6738
Reagent and conditions:
(a) (3R)-3-methylmorpholine, TEA, DCM, 77%;
(b) LiBH4, THF, 100%;
(c) MsCl, TEA, DCM, 100%;
(d) LiI, dioxane, 78%;
(e) NaSMe, DMF, 96%;
(f) m-CPBA, DCM;
(g) crystallization or chromatography, 40% (two steps);
AZD6738 is currently being tested in multiple phase I/II trials for the treatment of cancer. Its structure, comprising a pyrimidine core decorated with a chiral morpholine, a cyclopropyl sulfoximine, and an azaindole, make it a challenging molecule to synthesize on a large scale. We describe the evolution of the chemical processes, following the manufacture of AZD6738 from the initial scale-up through to multikilos on plant scale. During this evolution, we developed a biocatalytic process to install the sulfoxide with high enantioselectivity, followed by introduction of the cyclopropyl group first in batch, then in a continuous flow plate reactor, and finally through a series of continuous stirred tank reactors. The final plant scale process to form AZD6738 was operated on 46 kg scale with an overall yield of 18%. We discuss the impurities formed throughout the process and highlight the limitations of this route for further scale-up.
imino-methyl-[1-[6-[(3R)-3-methylmorpholin-4-yl]-2-(1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl]cyclopropyl]-oxo-λ6-sulfane (1) (30.0 g) were added at 75 °C, and the reaction mixture was held for 2 h. The mixture was cooled to 20 °C, and n-heptane (141.9 kg) was added at the rate of 40 kg/h. The solid was collected by filtration, washed with a mixture of 1-butanol and n-heptane (9.3 and 22.4 kg respectively), and then given a further wash with n-heptane (32.2 kg). The solid was dried at 40 °C to give imino-methyl-[1-[6-[(3R)-3-methylmorpholin-4-yl]-2-(1H-pyrrolo[2,3-b]pyridin-4-yl)pyrimidin-4-yl]cyclopropyl]-oxo-λ6-sulfane (1) as a whit solid (41.4 kg, 92% yield): Assay (HPLC) 99.9%; Assay (NMR) 99% wt/wt.
2: Wallez Y, Dunlop CR, Johnson TI, Koh SB, Fornari C, Yates JWT, Bernaldo de Quirós Fernández S, Lau A, Richards FM, Jodrell DI. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol Cancer Ther. 2018 Jun 11. doi: 10.1158/1535-7163.MCT-18-0010. [Epub ahead of print] PubMed PMID: 29891488.
3: Fròsina G, Profumo A, Marubbi D, Marcello D, Ravetti JL, Daga A. ATR kinase inhibitors NVP-BEZ235 and AZD6738 effectively penetrate the brain after systemic administration. Radiat Oncol. 2018 Apr 23;13(1):76. doi: 10.1186/s13014-018-1020-3. PubMed PMID: 29685176; PubMed Central PMCID: PMC5914052.
4: Zhang J, Dulak AM, Hattersley MM, Willis BS, Nikkilä J, Wang A, Lau A, Reimer C, Zinda M, Fawell SE, Mills GB, Chen H. BRD4 facilitates replication stress-induced DNA damage response. Oncogene. 2018 Jul;37(28):3763-3777. doi: 10.1038/s41388-018-0194-3. Epub 2018 Apr 11. PubMed PMID: 29636547.
5: Jin J, Fang H, Yang F, Ji W, Guan N, Sun Z, Shi Y, Zhou G, Guan X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia. 2018 May;20(5):478-488. doi: 10.1016/j.neo.2018.03.003. Epub 2018 Mar 30. PubMed PMID: 29605721; PubMed Central PMCID: PMC5915994.
6: Henssen AG, Reed C, Jiang E, Garcia HD, von Stebut J, MacArthur IC, Hundsdoerfer P, Kim JH, de Stanchina E, Kuwahara Y, Hosoi H, Ganem NJ, Dela Cruz F, Kung AL, Schulte JH, Petrini JH, Kentsis A. Therapeutic targeting of PGBD5-induced DNA repair dependency in pediatric solid tumors. Sci Transl Med. 2017 Nov 1;9(414). pii: eaam9078. doi: 10.1126/scitranslmed.aam9078. PubMed PMID: 29093183; PubMed Central PMCID: PMC5683417.
7: Jones BC, Markandu R, Gu C, Scarfe G. CYP-Mediated Sulfoximine Deimination of AZD6738. Drug Metab Dispos. 2017 Nov;45(11):1133-1138. doi: 10.1124/dmd.117.077776. Epub 2017 Aug 23. PubMed PMID: 28835442.
8: Dunne V, Ghita M, Small DM, Coffey CBM, Weldon S, Taggart CC, Osman SO, McGarry CK, Prise KM, Hanna GG, Butterworth KT. Inhibition of ataxia telangiectasia related-3 (ATR) improves therapeutic index in preclinical models of non-small cell lung cancer (NSCLC) radiotherapy. Radiother Oncol. 2017 Sep;124(3):475-481. doi: 10.1016/j.radonc.2017.06.025. Epub 2017 Jul 8. PubMed PMID: 28697853.
9: Kiesel BF, Shogan JC, Rachid M, Parise RA, Vendetti FP, Bakkenist CJ, Beumer JH. LC-MS/MS assay for the simultaneous quantitation of the ATM inhibitor AZ31 and the ATR inhibitor AZD6738 in mouse plasma. J Pharm Biomed Anal. 2017 May 10;138:158-165. doi: 10.1016/j.jpba.2017.01.055. Epub 2017 Feb 4. PubMed PMID: 28213176; PubMed Central PMCID: PMC5357441.
10: Ma J, Li X, Su Y, Zhao J, Luedtke DA, Epshteyn V, Edwards H, Wang G, Wang Z, Chu R, Taub JW, Lin H, Wang Y, Ge Y. Mechanisms responsible for the synergistic antileukemic interactions between ATR inhibition and cytarabine in acute myeloid leukemia cells. Sci Rep. 2017 Feb 8;7:41950. doi: 10.1038/srep41950. PubMed PMID: 28176818; PubMed Central PMCID: PMC5296912.
11: Vendetti FP, Leibowitz BJ, Barnes J, Schamus S, Kiesel BF, Abberbock S, Conrads T, Clump DA, Cadogan E, O’Connor MJ, Yu J, Beumer JH, Bakkenist CJ. Pharmacologic ATM but not ATR kinase inhibition abrogates p21-dependent G1 arrest and promotes gastrointestinal syndrome after total body irradiation. Sci Rep. 2017 Feb 1;7:41892. doi: 10.1038/srep41892. PubMed PMID: 28145510; PubMed Central PMCID: PMC5286430.
12: Min A, Im SA, Jang H, Kim S, Lee M, Kim DK, Yang Y, Kim HJ, Lee KH, Kim JW, Kim TY, Oh DY, Brown J, Lau A, O’Connor MJ, Bang YJ. AZD6738, A Novel Oral Inhibitor of ATR, Induces Synthetic Lethality with ATM Deficiency in Gastric Cancer Cells. Mol Cancer Ther. 2017 Apr;16(4):566-577. doi: 10.1158/1535-7163.MCT-16-0378. Epub 2017 Jan 30. PubMed PMID: 28138034.
13: Dillon MT, Barker HE, Pedersen M, Hafsi H, Bhide SA, Newbold KL, Nutting CM, McLaughlin M, Harrington KJ. Radiosensitization by the ATR Inhibitor AZD6738 through Generation of Acentric Micronuclei. Mol Cancer Ther. 2017 Jan;16(1):25-34. doi: 10.1158/1535-7163.MCT-16-0239. Epub 2016 Nov 9. PubMed PMID: 28062704; PubMed Central PMCID: PMC5302142.
14: Kim H, George E, Ragland R, Rafial S, Zhang R, Krepler C, Morgan M, Herlyn M, Brown E, Simpkins F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin Cancer Res. 2017 Jun 15;23(12):3097-3108. doi: 10.1158/1078-0432.CCR-16-2273. Epub 2016 Dec 19. PubMed PMID: 27993965; PubMed Central PMCID: PMC5474193.
15: Kim HJ, Min A, Im SA, Jang H, Lee KH, Lau A, Lee M, Kim S, Yang Y, Kim J, Kim TY, Oh DY, Brown J, O’Connor MJ, Bang YJ. Anti-tumor activity of the ATR inhibitor AZD6738 in HER2 positive breast cancer cells. Int J Cancer. 2017 Jan 1;140(1):109-119. doi: 10.1002/ijc.30373. Epub 2016 Oct 21. PubMed PMID: 27501113.
16: Biskup E, Naym DG, Gniadecki R. Small-molecule inhibitors of Ataxia Telangiectasia and Rad3 related kinase (ATR) sensitize lymphoma cells to UVA radiation. J Dermatol Sci. 2016 Dec;84(3):239-247. doi: 10.1016/j.jdermsci.2016.09.010. Epub 2016 Sep 16. PubMed PMID: 27743911.
17: Checkley S, MacCallum L, Yates J, Jasper P, Luo H, Tolsma J, Bendtsen C. Corrigendum: Bridging the gap between in vitro and in vivo: Dose and schedule predictions for the ATR inhibitor AZD6738. Sci Rep. 2016 Feb 9;6:16545. doi: 10.1038/srep16545. PubMed PMID: 26859465; PubMed Central PMCID: PMC4747154.
18: Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E, Stewart G, Brown J, Lau A, Pratt G, Parry H, Taylor M, Moss P, Hillmen P, Stankovic T. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood. 2016 Feb 4;127(5):582-95. doi: 10.1182/blood-2015-05-644872. Epub 2015 Nov 12. PubMed PMID: 26563132.
19: Vendetti FP, Lau A, Schamus S, Conrads TP, O’Connor MJ, Bakkenist CJ. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget. 2015 Dec 29;6(42):44289-305. doi: 10.18632/oncotarget.6247. PubMed PMID: 26517239; PubMed Central PMCID: PMC4792557.
20: Karnitz LM, Zou L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin Cancer Res. 2015 Nov 1;21(21):4780-5. doi: 10.1158/1078-0432.CCR-15-0479. Epub 2015 Sep 11. Review. PubMed PMID: 26362996; PubMed Central PMCID: PMC4631635.
//////AZD6738, AZD-6738, AZD 6738, AstraZeneca, University of Pennsylvania, Phase II, Breast cancer, Gastric cancer, Non-small cell lung cancer, Ovarian cancer, Ceralasertib
Selpercatinib is a tyrosine kinase inhibitor with antineoplastic properties.
A phase I/II trial is also under way in pediatric patients and young adults with activating RET alterations and advanced solid or primary CNS tumors.
Loxo Oncology (a wholly-owned subsidiary of Eli Lilly ), under license from Array , is developing selpercatinib, a lead from a program of RET kinase inhibitors, for treating cancer, including non-small-cell lung cancer, medullary thyroid cancer, colon cancer, breast cancer, pancreatic cancer, papillary thyroid cancer, other solid tumors, infantile myofibromatosis, infantile fibrosarcoma and soft tissue sarcoma
In 2018, the compound was granted orphan drug designation in the U.S. for the treatment of pancreatic cancer and in the E.U. for the treatment of medullary thyroid carcinoma.
Trk is a high affinity receptor tyrosine kinase activated by a group of soluble growth factors called neurotrophic factor (NT). The Trk receptor family has three members, namely TrkA, TrkB and TrkC. Among the neurotrophic factors are (1) nerve growth factor (NGF) which activates TrkA, (2) brain-derived neurotrophic factor (BDNF) and NT4/5 which activate TrkB, and (3) NT3 which activates TrkC. Trk is widely expressed in neuronal tissues and is involved in the maintenance, signaling and survival of neuronal cells.
The literature also shows that Trk overexpression, activation, amplification and/or mutations are associated with many cancers including neuroblastoma, ovarian cancer, breast cancer, prostate cancer, pancreatic cancer, multiple myeloma, astrocytoma. And medulloblastoma, glioma, melanoma, thyroid cancer, pancreatic cancer, large cell neuroendocrine tumor and colorectal cancer. In addition, inhibitors of the Trk/neurotrophin pathway have been shown to be effective in a variety of preclinical animal models for the treatment of pain and inflammatory diseases.
The neurotrophin/Trk pathway, particularly the BDNF/TrkB pathway, has also been implicated in the pathogenesis of neurodegenerative diseases, including multiple sclerosis, Parkinson’s disease, and Alzheimer’s disease. The modulating neurotrophic factor/Trk pathway can be used to treat these and related diseases.
It is believed that the TrkA receptor is critical for the disease process in the parasitic infection of Trypanosoma cruzi (Chagas disease) in human hosts. Therefore, TrkA inhibitors can be used to treat Chagas disease and related protozoal infections.
Trk inhibitors can also be used to treat diseases associated with imbalances in bone remodeling, such as osteoporosis, rheumatoid arthritis, and bone metastasis. Bone metastases are a common complication of cancer, up to 70% in patients with advanced breast or prostate cancer and about 15 in patients with lung, colon, stomach, bladder, uterine, rectal, thyroid or kidney cancer Up to 30%. Osteolytic metastases can cause severe pain, pathological fractures, life-threatening hypercalcemia, spinal cord compression, and other neurostress syndromes. For these reasons, bone metastases are a serious cancer complication that is costly. Therefore, an agent that can induce apoptosis of proliferating bone cells is very advantageous. Expression of the TrkA receptor and TrkC receptor has been observed in the osteogenic region of the fractured mouse model. In addition, almost all osteoblast apoptosis agents are very advantageous. Expression of the TrkA receptor and TrkC receptor has been observed in the osteogenic region of the fractured mouse model. In addition, localization of NGF was observed in almost all osteoblasts. Recently, it was demonstrated that pan-Trk inhibitors in human hFOB osteoblasts inhibit tyrosine signaling activated by neurotrophic factors that bind to all three Trk receptors. This data supports the theory of using Trk inhibitors to treat bone remodeling diseases, such as bone metastases in cancer patients.
Developed by Loxo Oncology, Larotrectinib (LOXO-101) is a broad-spectrum antineoplastic agent for all tumor patients expressing Trk, rather than tumors at an anatomical location. LOXO-101 chemical name is (S)-N-(5-((R)-2-(2,5-difluorophenyl)-pyrrolidin-1-yl)pyrazolo[1,5-a] Pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide, the structural formula is as follows. LOXO-101 began treatment of the first patient in March 2015; on July 13, 2016, the FDA granted a breakthrough drug qualification for the inoperable removal or metastatic solid tumor of adults and children with positive Trk fusion gene mutations; Key entry was completed in February 2017; in November 2018, the FDA approved the listing under the trade name Vitrakvi.
Poor absorption, distribution, metabolism, and/or excretion (ADME) properties are known to be the primary cause of clinical trial failure in many drug candidates. Many of the drugs currently on the market also limit their range of applications due to poor ADME properties. The rapid metabolism of drugs can lead to the inability of many drugs that could be effectively treated to treat diseases because they are too quickly removed from the body. Frequent or high-dose medications may solve the problem of rapid drug clearance, but this approach can lead to problems such as poor patient compliance, side effects caused by high-dose medications, and increased treatment costs. In addition, rapidly metabolizing drugs may also expose patients to undesirable toxic or reactive metabolites.
Although LOXO-101 is effective as a Trk inhibitor in the treatment of a variety of cancers and the like, it has been found that a novel compound having a good oral bioavailability and a drug-forming property for treating a cancer or the like is a challenging task. Thus, there remains a need in the art to develop compounds having selective inhibitory activity or better pharmacodynamics/pharmacokinetics for Trk kinase mediated diseases useful as therapeutic agents, and the present invention provides such compounds.
Compounds of Formula I-IV, 4-(6-(4-((6-methoxypyridin-3-yl)methyl)piperazin-1-yl)pyridin-3-yl)-6-(1-methyl-1H-pyrazol-4-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula I); 6-(2-hydroxy-2-methylpropoxy)-4-(6-(6-((6-methoxypyridin-3-yl)methyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula II); 6-(2-hydroxy-2-methylpropoxy)-4-(6-(6-(6-methoxynicotinoyl)-3,6-diazabicyclo[3.1.1]heptan-3-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula III); and 6-(2-hydroxy-2-methylpropoxy)-4-(6-(4-hydroxy-4-(pyridin-2-ylmethyl)piperidin-1-yl)pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile (Formula IV) are inhibitors of RET kinase, and are useful for treating diseases such as proliferative diseases, including cancers.
[0007] Accordingly, provided herein is a compound of Formula I-IV:
and pharmaceutically acceptable salts, amorphous, and polymorph forms thereof.
PATENT
WO 2019075114
PATENT
WO-2019120194
Novel deuterated analogs of pyrazolo[1,5-a]pyrimidine compounds, particularly selpercatinib , processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating pain, inflammation, cancer and certain infectious diseases.
Example 2(S)-N-(5-((R)-2-(2,5-difluorophenyl)pyrrolidin-1-yl-2,3,3-d 3)-pyrazolo[ 1,5-a] pyrimidin-3-yl) -3-hydroxypyrazole prepared pyrrolidine-1-carboxamide (compound L-2) a.
Axelar is developing picropodophyllin, a small-molecule IGF-1 receptor antagonist for the treatment of cancer including NSCLC and malignant astrocytoma. In February 2019, a phase Ia study was planned to initiate for solid tumor in March 2019.
Picropodophyllin is a cyclolignan alkaloid found in the mayapple plant family (Podophyllum peltatum), and a small molecule inhibitor of the insulin-like growth factor 1 receptor (IGF1R) with potential antineoplastic activity. Picropodophyllin specifically inhibits the activity and downregulates the cellular expression of IGF1R without interfering with activities of other growth factor receptors, such as receptors for insulin, epidermal growth factor, platelet-derived growth factor, fibroblast growth factor and mast/stem cell growth factor (KIT). This agent shows potent activity in the suppression o f tumor cell proliferation and the induction of tumor cell apoptosis. IGF1R, a receptor tyrosine kinase overexpressed in a variety of human cancers, plays a critical role in the growth and survival of many types of cancer cells.
Picropodophyllotoxin is an organic heterotetracyclic compound that has a furonaphthodioxole skeleton bearing 3,4,5-trimethoxyphenyl and hydroxy substituents. It has a role as an antineoplastic agent, a tyrosine kinase inhibitor, an insulin-like growth factor receptor 1 antagonist and a plant metabolite. It is a lignan, a furonaphthodioxole and an organic heterotetracyclic compound.
Picropodophyllin has been investigated for the treatment of Non Small Cell Lung Cancer.
One of the largest challenges in pharmaceutical drug development is that drug compounds often are poorly soluble, or even insoluble, in aqeous media. Insufficient drug solubility means insufficient bioavailability, as well as poor plasma exposure of the drug when administered to humans and animals. Variability of plasma exposure in humans is yet a problem when developing drugs which are poorly soluble, or even insoluble, in aqeous media.
It is estimated that between 40% and 70 % of all new chemical entities identified in drug discovery programs, are insufficiently soluble in aqeous media (M. Lindenberg, S et al: European Journal of Pharmaceutics and Biopharmaceuticals, vol. 58, no.2, pp. 265-278, 2004). Scientists have investigated various ways of solving the problem with poor drug solubility in order to enhance bioavailability of poorly absorbed drugs, aiming at increasing their clinical efficacy when administered orally.
Technologies such as increase of the surface area and hence dissolution may sometimes solve solubility problems. Other techniques that may also solve bioavailability problems are addition of surfactants and polymers. However, each chemical compound has its own unique chemical and physical properties, and hence has its own unique challenges when being formulated into a pharmaceutical product that can exert its clinical efficacy.
Picropodophyllin is an insulin-like growth factor-1 receptor inhibitor fiGF-lR inhibitor) small-molecule compound belonging to the class of compounds denominated cyclolignans, having the chemical structure:
The patent applicant is presently entering clinical phase II development with its development compound picropodophyllin (AXL1717). However, picropodophyllin is poorly soluble in aqueous media. In a phase I clinical study performed by the applicant in 2012 (Ekman S et al; Acta Oncologica, 2016; 55: pp. 140-148), it was discovered that picropodophyllin, when administered as an oral suspension to lung cancer patients, resulted in unacceptable variability in drug exposure. A large variability in plasma exposure of the active drug picropodophyllin occurred not only within certain patients, but also between several patients.
Yet a problem with administering picropodophyllin as an aqeous solution, is that due to the poor solubility in aqueous media, it is difficult or even impossible to reach the required therapeutic doses.
The compound picropodophyllin is furthermore physically unstable, and transforms from amorphous picropodophyllin into crystalline picropodophyllin. Yet a stability problem with picropodophyllin is that it is chemically unstable in solution.
Novel amorphous forms of picropodophyllin , processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating cancers, such as neurologic cancer, lung cancer, breast cancer, head and neck cancer, gastrointestinal cancer, genitourinary cancer, gynecologic cancer, hematologic cancer, musculoskeletal cancer, skin cancer, endocrine cancer, and eye cancers. , claiming picropodophyllin derivatives as modulators of insulin-like growth factor-1 receptor (IGF-1), useful for treating cancers, assigned to Axelar AB ,
A nickel-catalyzed reductive cascade approach to the efficient construction of diastereodivergent cores embedded in podophyllum lignans is developed for the first time. Their gram-scale access paved the way for unified syntheses of naturally occurring podophyllotoxin and other members.
he first catalytic enantioselective total synthesis of (−)-podophyllotoxin is accomplished by a challenging organocatalytic cross-aldol Heck cyclization and distal stereocontrolled transfer hydrogenation in five steps from three aldehydes. Reversal of selectivity in hydrogenation led to the syntheses of other stereoisomers from the common precursor.
(-)-Picropodophyllin 4. The lactone 5 (0.2 g, 0.38 mmol) was taken in 1-pentanol (5 mL) in a double neck RB flask at rt. Water (0.14 mL, 7.6 mmol) was added to above mixture and it was then degassed with argon followed by addition of Pd/C (0.04 g, 20% by wt.) and HCO2Na (0.78g, 11.4 mmol). The reaction mixture was heated at 40 °C for 12 h. On completion, the reaction mixture was diluted with EtOAc (200 mL), filtered through a celite pad and solvent was removed under vacuum. This crude mixture was dissolved in THF (3.8 mL), TBAF (1.9 mL, 1.9 mmol, 1M in THF) was added and stirred for 6 h at 27 °C. On completion, EtOAc (250 mL) was added, washed with water (100 mL), brine and dried over Na2SO4. After removal of solvent, the crude product was purified by column chromatography (hexanes-EtOAc, 3:2) to get the title compound as a white solid (0.082 g, 52%): Rf 0.32 (hexanes/EtOAc, 1:1); [α]25 D = -10.6 (c = 0.4, CHCl3) [lit. -10 (c = 0.3, CHCl3), -11 (c = 0.41, CHCl3)]3a,b;
The formylation of 6-bromo-1,3-benzodioxole-5-carbaldehyde dimethyl acetal (I) with BuLi and DMF gives the 6-formyl derivative (II), which is reduced with NaBH4 in ethanol to yield the corresponding carbinol (III). The cyclization of (III) with dimethyl acetylenedicarboxylate (V) in hot acetic acid (through the nonisolated intermediate (IV)) affords dimethyl 1,4-epoxy-6,7-(methylenedioxy)naphthalene-2,3-dicarboxylate (VI), which is hydrogenated with H2 over Pd/C in ethyl acetate to give the (1R*,2S*,3R*,4S*)-tetrahydro derivative (VII). The reduction of (VII) with LiAlH4 in refluxing ethyl ether affords the corresponding bis carbinol (VIII), which is treated with acetic anhydride to afford the diacetate (IX). The enzymatic monodeacetylation of (VIII) with PPL enzyme in DMSO/buffer gives (1R,2R,3S,4S)-2-(acetoxymethyl)-1,4-epoxy-3-(hydroxymethyl)-6,7-(methylenedioxy)-1,2,3,4-tetrahydronaphthalene (X), which is silylated with TBDMS-Cl and imidazole in DMF yielding the silyl ether (XI). The hydrolysis of the acetoxy group of (XI) with K2CO3 in methanol affords the carbinol (XII), which is oxidized with oxalyl chloride in dichloromethane affording the carbaldehyde (XIII). The exchange of the silyl protecting group of (XIII) (for stability problems) provided the triisopropylsilyl ether (XIV), which is treated with sodium methoxide in methanol to open the epoxide ring yielding the hydroxy aldehyde (XV). The protection of the hydroxy group of (XV) with 2-(trimethylsilyl)ethoxymethyl chloride and DIEA in dichloromethane provides the corresponding ether (XVI). The carbinol (III) can also be obtained directly from 6-bromo-1,3-benzodioxole-5-carbaldehyde dimethyl acetal (I) by reaction with formaldehyde and BuLi in THF.
The oxidation of the aldehyde group of (XVI) with NaClO2 in tert-butanol affords the corresponding carboxylic acid (XVII), which is condensed with 2-oxazolidinone (XVIII) by means of carbonyldiimidazole (CDI) in THF to give the acyl imidazolide (XIX). The arylation of (XIX) with 3,4,5-trimethoxyphenylmagnesium bromide (XX) in THF yields the expected addition product (XXI), which is cyclized by means of TBAF in hot THF to afford the tetracyclic intermediate (XXII). Isomerization of the cis-lactone ring of (XXII) with LDA in THF affords intermediate (XXIII) with its lactone ring with the correct trans-conformation. Finally, this compound is deprotected with ethyl mercaptane and MgBr2 in ethyl ether to provide the target compound.
Synthesis 1992,719
The intermediate trans-8-oxo-5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetra-hydronaphtho[2,3-d][1,3]benzodioxole-6-carboxylic acid ethyl ester (XI) has been obtained by several different ways: (a) The condensation of benzophenone (XXXVIII) with diethyl malonate (XXXIX) by means of t-BuOK gives the alkylidenemalonate (XL), which is hydrogenated with H2 over Pd/C to the alkylmalonate hemiester (XLI). The reaction of (XLI) with acetyl chloride affords the mixed anhydride (XLII), which is finally cyclized to the target (XI) by means of SnCl4. (b) The cyclization of the malonic ester derivative (XLIII) by means of Ti(CF3–CO2)3 gives the 5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydronaphtho [2,3-d][1,3]dioxole-6,6-dicarboxylic acid dimethyl ester (XLIV), which is finally oxidized and decarboxylated with NBS and NaOH in methanol to afford the target intermediate (XI). (c) The cyclization of the benzylidenemalonate (XLV) with the aryllithium derivative (XLVI) gives the 8-methoxy-5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydronaphtho[2,3-d][1,3]dioxole-6,6-dicarboxylic acid dimethyl ester (XLVII), which is demethylated with TFA and oxidized with CrO3 and pyridine to the target compound (XI). (d) The cyclopropanation of the chalcone (XLVIII) with (ethoxycarbonyl) (dimethylsulfonium)methylide (XLIX) gives the cyclopropanecarboxylate (L), which is finally rearranged with BF3/Et2O to the target intermediate (IX).
The cyclization of 3,4,5-trimethoxycinnamic acid ethyl ester (LI) with malonic acid ethyl ester potassium salt (LII) by means of Mn(OAc)3 gives the tetrahydrofuranone (LIII), which is acylated with 1,3-benzodioxol-5-ylcarbonyl chloride (LIV) yielding the tetrahydrofuranone (LV). Finally, this compound is rearranged and decarboxylated with SnCl4 to the target intermediate (XI).
The cyclization of 6-[1-hydroxy-1-(3,4,5-trimethoxyphenyl)methyl]-1,3-benzodioxol-5-carbaldehyde dimethylacetal (LVI) by means of AcOH gives 5-(3,4,5-trimethoxyphenyl)-1,3-dioxolo[4,5-f]isobenzofuran (LVII), which is submitted to a Diels-Alder cyclization with acetylenedicarboxylic acid dimethyl ester (LVIII) yielding the epoxy derivative (LIX). The selective reduction of (LIX) with LiBEt3H and H2 affords the carbinol (LX), which is treated with H2 over RaNi in order to open the epoxide ring to give the diol (LXI) with the wrong configuration at the secondary OH group. The treatment of (LXI) with aqueous acid isomerizes the secondary OH group to (LXII) with the suitable configuration. Finally, this compound is cyclized with DCC to the desired target compound.
The Diels-Alder cyclization of 5-(3,4,5-trimethoxyphenyl)-7H-pyrano[3,4-f][1,3]benzodioxol-7-one (I) with dimethyl maleate (LXIII) gives the expected adduct (LXIV), which by thermal extrusion of CO2 yields the dihydronaphthodioxole (LXV). This compound is then converted to dihydroxycompound (X), which is finally cyclized by means of ZnCl2 to provide the target compound. The Diels-Alder cyclization of 5-(3,4,5-trimethoxyphenyl)-7H-pyrano[3,4-f][1,3]benzodioxol-7-one (I) with dimethyl fumarate (LXVI) gives the expected adduct (LXVII), which by hydrogenation with H2 over Pd/C yields the tricarboxylic acid derivative (LXVIII). The reaction of (LXVIII) with Pb(OAc)4 affords the acetoxy derivative (LXIX), which is selectively reduced with LiBEt3H providing the diol (LXI) with the wrong configuration at the secondary OH group. The treatment of (LXI) with aqueous acid isomerizes the secondary OH group to give the previously described (X) with the suitable configuration.
The reaction of benzocyclobutane derivative (LXX) with isocyanate (LXXI) by means of Ph3SnOAc gives the carbamate (LXXII), which is cyclized by a thermal treatment with LiOH yielding the tetracyclic carboxylic acid (LXXIII). The opening of the oxazinone ring of (LXXIII) in basic medium affords the tricyclic amino acid (LXXIV), which is finally cyclized to the target compound by reaction with sodium nitrite in acidic medium (pH = 4).
J Chem Soc Chem Commun 1993,1200
The Diels-Alder cyclization of 5-(3,4,5-trimethoxyphenyl)-7H-pyrano[3,4-f][1,3]benzodioxol-7-one (I) with the chiral dihydrofuranone (II) in hot acetonitrile gives the pentacyclic anhydride (III), which is opened with warm acetic acid yielding the carboxylic acid (IV). Hydrogenation of the benzylic double bond of (IV) with H2 over Pd/C affords (V), which is treated with lead tetraacetate and acetic acid in THF to give the acetoxy compound (VI). The hydrolysis of the acetoxy group and the menthol hemiacetal group with HCl in hot dioxane yields the diol (VII), which is treated with diazomethane in ether/methanol affording the aldehyde (VIII). The reduction of the aldehyde group of (VIII) with LiEt3BH in THF gives the diol (IX) as a diastereomeric mixture, which is treated with HCl in THF to afford the diol (X) with the right conformation. Finally, this compound is lactonized to the target compound with ZnCl2 in THF.
Selinexor is an orally available, small molecule inhibitor of CRM1 (chromosome region maintenance 1 protein, exportin 1 or XPO1), with potential antineoplastic activity. Selinexor modifies the essential CRM1-cargo binding residue cysteine-528, thereby irreversibly inactivates CRM1-mediated nuclear export of cargo proteins such as tumor suppressor proteins (TSPs), including p53, p21, BRCA1/2, pRB, FOXO, and other growth regulatory proteins. As a result, this agent, via the approach of selective inhibition of nuclear export (SINE), restores endogenous tumor suppressing processes to selectively eliminate tumor cells while sparing normal cells. CRM1, the major export factor for proteins from the nucleus to the cytoplasm, is overexpressed in a variety of cancer cell types.
Selinexor has been used in trials studying the treatment of AML, Glioma, Sarcoma, Leukemia, and Advanced, among others.
Selinexor, also known as KPT-330, is an orally bioavailable, potent and selective XPO1/CRM1 Inhibitor. Selinexor is effective in acquired resistance to ibrutinib and synergizes with ibrutinib in chronic lymphocytic leukemia. Selinexor potentiates the antitumor activity of gemcitabine in human pancreatic cancer through inhibition of tumor growth, depletion of the antiapoptotic proteins, and induction of apoptosis. Selinexor has strong activity against primary AML cells while sparing normal stem and progenitor cells.
SYN
Medical uses
Selinexor is restricted for use in combination with the steroid dexamethasone in people with relapsed or refractory multiple myelomawhich has failed to respond to at least four or five other therapies (so-called “quad-refractory” or “penta-refractory” myeloma),[5] for whom no other treatment options are available.[3][4] It is the first drug to be approved for this indication.[6]
Adverse effects
In the clinical study used to support FDA approval, selinexor was associated with high rates of pancytopenia, including leukopenia(28%), neutropenia (34%, severe in 21%), thrombocytopenia (74%, severe in 61% of patients), and anemia (59%).[4][7] The most common non-hematological side effects were gastrointestinal reactions (nausea, anorexia, vomiting, and diarrhea), hyponatremia (low blood sodium levels, occurring in up to 40% of patients), and fatigue.[7][8] More than half of all patients who received the drug developed infections, including fatal cases of sepsis.[7] However, these data are from an open-label trial, and thus cannot be compared to placebo or directly attributed to treatment.
Mechanism of action
Schematic illustration of the Ran cycle of nuclear transport. Selinexor inhibits this process at the nuclear export receptor (upper right).
Like other so-called selective inhibitors of nuclear export (SINEs), selinexor works by binding to exportin 1 (also known as CRM1). CRM1 is a karyopherin which performs nuclear transport of several proteins, including tumor suppressors, oncogenes, and proteins involved in governing cell growth, from the cell nucleus to the cytoplasm; it is often overexpressed and its function misregulated in several types of cancer.[1] By restoring nuclear transport of these proteins to normal, SINEs lead to a buildup of tumor suppressors in the nucleus of malignant cells and reduce levels of oncogene products which drive cell proliferation. This ultimately leads to cell cycle arrest and death of cancer cells by apoptosis.[1][2][7]In vitro, this effect appeared to spare normal (non-malignant) cells.[1][8]
Because CRM1 is a pleiotropicgene, inhibiting it affects many different systems in the body, which explains the high incidence of adverse reactions to selinexor.[2] Thrombocytopenia, for example, is a mechanistic and dose-dependent effect, occurring because selinexor causes a buildup of the transcription factor STAT3 in the nucleus of hematopoietic stem cells, preventing their differentiation into mature megakaryocytes (platelet-producing cells) and thus slowing production of new platelets.[2]
Selinexor was developed by Karyopharm Therapeutics of Newton, Massachusetts, a pharmaceutical company devoted entirely to the development of drugs that target nuclear transport. It was approved by the FDA on July 3, 2019, on the basis of a single uncontrolled clinical trial. The decision was controversial, and overruled the previous recommendation of an FDA Advisory Panel which had voted 8–5 against approving the drug, due to concerns about efficacy and toxicity.[3]
As of 2019, phase I/II and III trials are ongoing,[3][9] including the use of selinexor in other cancers and in combinations with other drugs used for multiple myeloma.[2]
International Publication No. WO 2013/019548 describes a series of compounds that are indicated to have inhibitory activity against chromosomal region maintenance 1 (CRM1, also referred to as exportin 1 or XPO1) and to be useful in the treatment of disorders associated with CRM1 activity, such as cancer. (Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-1H-1,2,4-triazol-1-yl)-N’-(pyrazin-2-yl)acrylohydrazide (also referred to as selinexor) is one of the compounds disclosed in International Publication No. WO 2013/019548. Selinexor has the chemical structure shown in Structural Formula I:
Example 1. Preparation of Selinexor Lot No.1305365 (Form A).
[00274] Selinexor for Lot No. 1305365 was made in accordance with the following reaction scheme:
[00275] A solution of propane phosphonic acid anhydride (T3P®, 50% in ethyl acetate, 35Kg) in THF (24.6Kg) was cooled to about -40 °C. To this solution was added a solution of KG1 (13.8Kg) and diisopropylethylamine (12.4Kg) in tetrahydrofuran (THF, 24.6Kg). The resulting mixture was stirred at about -40°C for approximately 2.5 hours.
[00276] In a separate vessel, KJ8 (4.80Kg) was mixed with THF (122.7Kg), and the resulting mixture cooled to about -20°C. The cold activated ester solution was then added to the KJ8 mixture with stirring, and the reaction was maintained at about -20°C. The mixture was warmed to about 5°C, water (138.1Kg) was added and the temperature adjusted to about 20°C. After agitating for about an hour, the lower phase was allowed to separate from the mixture and discarded. The upper layer was diluted with ethyl acetate (EtOAc). The organic phase was then washed three times with potassium phosphate dibasic solution (~150Kg), then with water (138.6Kg).
[00277] The resulting organic solution was concentrated under reduced pressure to 95L, EtOAc (186.6Kg) was added and the distillation repeated to a volume of 90L. Additional EtOAc (186.8Kg) was added and the distillation repeated a third time to a volume of 90L. The batch was filtered to clarify, further distilled to 70L, then heated to about 75°C, and slowly cooled to 0 to 5°C. The resulting slurry was filtered and the filter cake washed with a mixture of EtOAc (6.3Kg) and toluene (17.9Kg) before being dried in a vacuum oven to provide selinexor designated Lot No. 1305365 (Form A).
Example 2. Preparation of Selinexor Lot No.1341-AK-109-2 (Form A).
[00278] The acetonitrile solvate of selinexor was prepared in accordance with Example 6.
[00279] The acetonitrile solvate of selinexor (2.7g) was suspended in a mixture of isopropanol (IPA, 8mL) and water (8mL), and the resulting mixture heated to 65 to 70 °C to effect dissolution. The solution was cooled to 45 °C, and water (28mL) was added over 15 minutes, maintaining the temperature between 40 and 45 °C. The slurry was cooled to 20 to 25 °C over an hour, then further cooled to 0 to 5 °C and held at that temperature for 30 minutes before being filtered. The filter cake was washed with 20% v/v IPA in water and the product dried under suction overnight, then in vacuo (40°C).
Example 3. Preparation of SelinexorSelinexorSelinexor Lot No. PC-14-005 (Form A).
[00280] The acetonitrile solvate of selinexor (Form D) was prepared in accordance with the procedure described in Example 6.
[00281] The acetonitrile solvate of selinexor (1.07Kg) was suspended in a mixture of IPA (2.52Kg) and water (3.2Kg) and the mixture heated to 70 to 75 °C to dissolve. The temperature was then adjusted to 40 to 45 °C and held at that temperature for 30 minutes. Water (10.7Kg) was added while maintaining the temperature at 40 to 45 °C, then the batch was cooled to 20 to 25 °C and agitated at that temperature for 4 hours before being further cooled to 0 to 5 °C. After a further hour of agitation, the slurry was filtered and the filter cake washed with a cold mixture of IPA (0.84Kg) and water (4.28Kg) before being dried.
Example 4. Preparation of SelinexorSelinexorSelinexor Lot No. PC-14-009 (Form A).
[00282] The acetonitrile solvate of selinexor (Form D) was prepared in accordance with the procedure described in Example 6.
[00283] The acetonitrile solvate of selinexor (1.5Kg) was suspended in IPA (3.6Kg) and water (4.5Kg) and warmed to 37 to 42 °C with gentle agitation. The suspension was agitated at that temperature for 4 hours, and was then cooled to 15 to 20 °C over 1 hour. Water (15.1Kg) was added, maintaining the temperature, then the agitation was continued for 1 hour and the batch was filtered. The filter cake was washed with a mixture of IPA (1.2Kg) and water (6Kg), then dried under a flow of nitrogen.
Example 5. Preparation of Selinexor Lot Nos.1339-BS-142-1, 1339-BS-142-2 and PC-14-008 (Form A).
[00284] A reactor, under nitrogen, was charged with KG1 (1Kg, 1.0 Eq), KJ8 (0.439 Kg, 1.4 Eq) and MeTHF (7L, 7 parts with respect to KG1). Diisopropylethylamine (0.902Kg, 2.45 Eq with respect to KG1) was added to the reaction mixture at -20 °C to -25 °C with a MeTHF rinse. To the reaction mixture, 50% T3P® in ethyl acetate (2.174Kg, 1.2 Eq with respect to KG1) was then charged, maintaining the temperature at -20 °C to -25 °C with a MeTHF rinse. After the completion of the addition, the reaction mixture was stirred briefly
and then warmed to 20 °C to 25 °C. Upon completion, the reaction mixture was washed first with water (5L, 5 parts with respect to KG1) and then with dilute brine (5L, 5 parts with respect to KG1). The organic layer was concentrated by vacuum distillation to a volume of 5 L (5 parts with respect to KG1), diluted with acetonitrile (15L, 15 parts with respect to KG1) at approximately 40 °C and concentrated again (5L, 5 parts with respect to KG1). After solvent exchange to acetonitrile, the reaction mixture was then heated to approximately 60 °C to obtain a clear solution. The reaction mixture was then cooled slowly to 0-5 °C, held briefly and filtered. The filter cake was washed with cold acetonitrile (2L, 5 parts with respect to KG1) and the filter cake was then dried under a stream of nitrogen to provide the acetonitrile solvate of selinexor (Form D) as a slightly off-white solid.
[00285] Form D of selinexor (0.9Kg) was suspended in IPA (2.1Kg, 2.7L, 3 parts with respect to Form D) and water (2.7Kg, 2.7L, 3 parts with respect to Form D) and warmed to approximately 40 °C. The resulting suspension was agitated for about 4 hours, selinexor, cooled to approximately 20 °C, and diluted with additional water (9Kg, 10 parts with respect to Form D). The mixture was stirred for a further 4-6 hours, then filtered, and the cake washed with a mixture of 20% IPA and water (4.5L, 5 parts with respect to Form D). The filter cake was then dried under vacuum to provide selinexor designated Lot No. PC-14-008 as a white crystalline powder with a >99.5% a/a UPLC purity (a/a=area to area of all peaks; UPLC-ultra performance HPLC).
Example 6. Preparation of Selinexor Lot No.1405463 (Form A).
[00286] Selinexor Lot No. 1405463 was prepared in accordance with the following reaction scheme:
.
[00287] A reactor was charged with KG1 (15.8Kg), KJ8 (6.9Kg) and MeTHF (90Kg). Diisopropylethylamine (14.2Kg) was added to the reaction mixture over approximately 35 minutes at about -20 °C. Following the addition of the diisopropylethylamine, T3P® (50%
solution in EtAOc, 34.4Kg) was added maintaining the temperature at -20 °C. The mixture stirred to complete the reaction first at -20 °C, then at ambient temperature.
[00288] Upon completion of the reaction, water (79Kg) was added over about 1 hour. The layers were separated and the organic layer was washed with a mixture of water (55Kg) and brine (18Kg), The mixture was filtered, and the methyl-THF/ethyl acetate in the mixture distillatively replaced with acetonitrile (volume of approximately 220L). The mixture was warmed to dissolve the solids, then slowly cooled to 0 to 5 °C before being filtered. The filter cake was washed with acetonitrile to provide the acetonitrile solvate of
selinexorSelinexorSelinexor (Form D).
[00289] The acetonitrile solvate of selinexorSelinexorSelinexor was dried, then mixed with isopropanol (23Kg) and water (55Kg). The slurry was warmed to about 38 °C and held at that temperature for approximately 4 hours before being cooled to 15 to 20 °C. Water (182Kg) was added. After a further 5 hours of agitation, the mixture was filtered and the filter cake washed with a mixture of isopropanol (14Kg) and water (73Kg), before being dried under vacuum (45 °C). The dried product was packaged to provide
selinexorSelinexorSelinexor Lot No. 1405463 (Form A).
Example 7. Polymorphism Studies of Selinexor.
[00290] A comprehensive polymorphism assessment of selinexor was performed in a range of different solvents, solvent mixtures and under a number of experimental conditions based on the solubility of selinexor. Three anhydrous polymorphs of
selinexorSelinexorSelinexor were observed by XRPD investigation, designated Form A, Form B and Form C. Form A is a highly crystalline, high-melting form, having a melting point of 177 °C, and was observed to be stable from a physico-chemical point of view when exposed for 4 weeks to 25 °C/97% relative humidity (RH) and to 40 °C/75% RH. A solvated form of selinexor was also observed in acetonitrile, designated Form D. A competitive slurry experiment confirmed Form A as the stable anhydrous form under the conditions investigated, except in acetonitrile, in which solvate formation was observed. It was further found that in acetonitrile, below 50 °C, only Form D is observed, at 50 °C both Form A and Form D are observed, and at 55 °C, Form A is observed .
Selinexor is an orally bioavailable selective nuclear export inhibitors, 2012 for the first time in clinical, so far carried out a total of 21 trials, indications include chronic myelogenous leukemia, acute myelogenous leukemia, acute lymphatic leukemia, prostate cancer, melanoma, non-small cell lung cancer, glioma, neuroblastoma into, gynecological cancer, diffuse large B-cell lymphoma, squamous cell carcinoma, colorectal cancer and the like.May 2014, FDA granted orphan drug designation Selinexor treatment of acute myeloid leukemia and diffuse large B-cell lymphoma, in June 2014, EMA is also granted orphan drug designation Selinexor treatment of both diseases.January 2015, received FDA orphan drug to treat multiple myeloma identified.
[0003] Currently, the synthesis process has been disclosed, the following reaction equation:
[0006] wherein the compound is 5 Selinexor drug.
[0007] In this method, however, easy to produce Intermediate 1-2 double bond is easily reversed when synthetically produced from trans impurities, in addition to more difficult to impact yield; Intermediate 3 Intermediate 4 Synthesis APIs 5 when required ultra-low temperature, and the product was purified by column required, only a yield of 20%.
SUMMARY
[0008] The object of the present invention to provide a novel compound Selinexor drug synthesis of 5, in order to solve technical problems.
[0009] – novel synthetic method of Se species I inexor drug, comprising the steps of:
Synthesis [0010] A, Compound 7
[0011] Compound 6, dichloromethane and ethyl acetate mixture, stirred and dissolved, compound 4, T3P (n-propyl phosphoric anhydride) and DIPEA (N, N- diisopropylethylamine) at a low temperature; the reaction was stirred for 25-35min at a low temperature, dichloromethane and water were added after the completion of the reaction, liquid separation, the organic phase was evaporated to dryness to give crude compound 7, crude without purification cast down;
[0012] B, Synthesis of Compound 8
[0013] the compound obtained in Step 7, and mixed sodium iodide acetic acid, warmed to 110-120 ° C, the reaction 2.5-3.5h; After completion of the reaction, the system cooled to room temperature, water and dichloromethane were added, stirred for 8 after -15min, standing layered organic phase was washed with saturated sodium bicarbonate and saturated sodium chloride, dried over anhydrous sodium sulfate and distilled to give crude compound 8, was dissolved in DMF (dimethyl fumarate) to give compound in DMF 8;
Synthesis [0014] C, of Compound 5
[0015] Compound 1, DBAC0 (triethylenediamine), the DMF mixed and dissolved with stirring, dropwise adding to the reaction system of the compound obtained in DMF step 8, after the addition was complete, stirring was continued for 3-4 hours; the reaction after completion, water and ethyl acetate were added to the system, the organic phase is evaporated to dryness and petroleum ether and recrystallized from ethyl acetate to give compound 5.
[0016] Preferably, said step A, the low temperature is 0-2 ° C.
[0017] Preferably, said step B in DMF, the crude compound 8 concentration of less than 1%.
[0018] The novel synthetic methods of the present invention Selinexor drug, the chemical equation is as follows:
〇
[0020] The present invention has the following advantages: novel synthetic method Selinexor drug of the present invention to overcome the conventional synthesis process, is easy to produce trans impurities, more difficult in addition, the influence the yield and the need for ultra-low temperature, and the product requires problems purified by column, the yield is very low, reducing the synthetic steps, increased yield, there is provided a new process for the synthesis of the drug Selinexor.
[0021] In addition to the above-described objects, features and advantages of the present invention as well as other objects, features and advantages.Below the invention will be described in further detail present.
Example 1
[0024] – novel synthetic method of Se species I inexor drug, comprising the steps of:
Synthesis [0025] A, Compound 7
[0026] 50ml three □ flask, 15ml of dichloromethane and 0.2g compound 6,15ml ethyl acetate, stirred and dissolved, was added 0.3g of compound 4 and 3gT3P, 0.75gDIPEA at 0 ° C; the system at 0 ° C the reaction was stirred for 30min, 50ml of dichloromethane and 30ml of water were added after the completion of the reaction, liquid separation, the organic phase was evaporated to dryness to give crude compound 7, crude without purification cast down;
[0027] B, Synthesis of Compound 8
[0028] 50ml three-necked flask, added the compound obtained in Step 7,40ml of glacial acetic acid and 1.38g of sodium iodide was heated to 115.(:, The reaction 3H; After completion of the reaction, cooled to room temperature system, the system will be transferred to 500ml flask, 50ml of water was added and IOOml dichloromethane, after stirring IOmin, standing separation, the organic phase was washed with saturated sodium bicarbonate and saturated washed with sodium chloride, dried over anhydrous sodium sulfate and distilled to give crude compound 8, was dissolved in IOmL DMF to give DMF solution of compound 8;
Synthesis [0029] C, of Compound 5
[0030] After 50ml 3-necked flask was added 0.2g compound 1,0.24gDBAC0,20mlDMF, dissolved with stirring, dropwise adding to the reaction system in DMF compound obtained in Step 8, after the addition was complete, stirring continued for 3.5 hours; after completion of the reaction, 20ml water was added to the system and 50ml ethyl acetate, the organic phase is evaporated to dryness and petroleum ether to ethyl acetate to give 0.158g of compound 5, yield 50.9%.
[0031] Example 2
[0032] – new type Se Iinexor drug synthesis, comprising the steps of:
Synthesis [0033] A, Compound 7
[0034] 50ml three □ flask, 15ml of dichloromethane and 0.2g compound 6,15ml ethyl acetate, stirred and dissolved, was added 0.3g of compound 4 and 3gT3P, 0.75gDIPEA at 1 ° C; system at 1 ° C the reaction was stirred for 35min, 50ml of dichloromethane and 30ml of water were added after the completion of the reaction, liquid separation, the organic phase was evaporated to dryness to give crude compound 7, crude without purification cast down;
[0035] B, Synthesis of Compound 8
Three-neck flask [0036] 50ml of addition of the compound obtained in Step 7,40ml glacial acetic acid and 1.38g of sodium iodide was heated to 120.(:, The reaction for 2.5 h; After completion of the reaction, cooled to room temperature system, the system will be transferred to 500ml flask, 60ml water and 120ml dichloromethane was added, after stirring for 15min, allowed to stand for separation, the organic phase was washed with saturated sodium bicarbonate and washed with saturated sodium chloride, dried over anhydrous sodium sulfate and distilled to give crude compound 8, 12mLDMF was dissolved in DMF to give a solution of compound 8;
Synthesis [0037] C, of Compound 5
[0038] After 50ml 3-necked flask was added 0.2g compound 1,0.24gDBAC0,20mlDMF, dissolved with stirring, dropwise adding to the reaction system of the compound obtained in DMF step 8, after the addition was complete, stirring continued for 3 hours; after completion of the reaction, 25ml of water and 50ml of ethyl acetate was added to the system, the organic phase is evaporated to dryness and petroleum ether to ethyl acetate to give 0.152g of compound 5, yield 49.0% billion
[0039] Example 3
[0040] – novel synthetic method of Se species I inexor drug, comprising the steps of:
Synthesis [0041] A, Compound 7
Three [0042] 50ml of flask, 15ml of dichloromethane and 0.2g compound 6,15ml ethyl acetate, stirred and dissolved, was added 0.3g of compound 4 and 3gT3P, 0.75gDIPEA at 2 ° C; system from 0 ° C the reaction was stirred for 25min, 40ml of dichloromethane and 35ml of water were added after the completion of the reaction, liquid separation, the organic phase was evaporated to dryness to give crude compound 7, crude without purification cast down;
[0043] B, Synthesis of Compound 8
Three-neck flask [0044] 50ml of addition of the compound obtained in Step 7,35ml glacial acetic acid and 1.38g of sodium iodide was heated to 110.(:, The reaction for 3.5 h; After completion of the reaction, cooled to room temperature system, the system will be transferred to 500ml flask, 50ml of water was added and dichloromethane IOOml After Smin of stirring, standing separation, the organic phase was washed with saturated sodium bicarbonate and washed with saturated sodium chloride, dried over anhydrous sodium sulfate and distilled to give crude compound 8, was dissolved in IOmL DMF to give DMF solution of compound 8;
Synthesis [0045] C, of Compound 5
[0046] 50ml three-neck flask was added 0.2g compound 1,0.24gDBA⑶, 20mlDMF, and dissolved with stirring, dropwise adding to the reaction system of the compound obtained in DMF step 8, after the addition was complete, stirring was continued for 4 hours; after completion of the reaction, 20ml of water and 40ml ethyl acetate were added to the system, the organic phase is evaporated to dryness and petroleum ether to ethyl acetate to give 0.155g of compound 5, yield 49.9% billion
The drug compound having the adopted name “Selinexor” has chemical name:(Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-IH-l,2,4-triazol-1 -yl)-N’-(pyrazin-2yl) acrylohydrazide as below.
Selinexor (KPT-330) is a first-in-class, oral Selective Inhibitor of Nuclear Export / SINE compound. Selinexor functions by binding with and inhibiting the nuclear export protein XP01 (also called CRM1 ), leading to the accumulation of tumor suppressor proteins in the cell nucleus. This reinitiates and amplifies their tumor suppressor function and is believed to lead to the selective induction of apoptosis in cancer cells, while largely sparing normal cells. Over 1 ,200 patients have been treated with Selinexor in company and investigator-sponsored Phase 1 and Phase 2 clinical trials in advanced hematologic malignancies and solid tumors. Karyopharm has initiated four later-phase clinical trials of Selinexor, including one in older patients with acute myeloid leukemia (SOPRA), one in patients with Richter’s transformation (SIRRT), one in patients with diffuse large B-cell lymphoma (SADAL) and a single-arm trial of Selinexor and lose-dose dexamethasone in patients with multiple myeloma (STORM). Patients may receive a twice-weekly combination of Selinexor in combination with low dose dexamethasone. Randomized 1 :1 , Selinexor will be dosed either at 60mg + dexamethasone or at 100 mg + dexamethasone.
US 8999996 B2 discloses Selinexor and a pharmaceutically acceptable salt thereof, pharmaceutical compositions and use for treating disorders associated with CRM1 activity. Further, it discloses preparative methods for the preparation of compounds disclosed therein including Selinexor by reacting (Z)-3-(3- (3,5-
bis(trifluoromethyl)phenyl)-IH-l,2,4-triazol-l-yl)acrylic acid in 1 :1 CH2CI2: AcOEt with 2-Hydrazinopyrazine at -40 °C followed by addition of T3P[Propylphosphonic anhydride] (50%) and DIPEA. After 30 minutes, the reaction mixture was concentrated and the crude oil was purified by preparative TLC using 5% MeOH in CH2CI2 as mobile phase (under ammonia atmosphere) to afford 40 mg of Selinexor with purity: 95.78%. However, it is not disclosed about the nature of the compound obtained therein.
WO 2016025904 A1 discloses various crystalline forms of Selinexor namely Form A, Form B, Form C, Form D, compositions and MoU thereof for the treatment of disorder associated with CRM1 activity and their preparative processes.
Prior art process for the preparation of Selinexor suffers from disadvantages interms of process such as the use of lengthy procedures to practice and resulting in low yields, which may not be viable at industrial scale. Synthetic product obtained therein has very low purity and contains significant amounts of unreacted starting materials and trans-isomer of Selinexor, which are further purified by time consuming and expensive chromatographic separations leading to loss of yield. Hence, there remains a need for improved process for the preparation of Selinexor which is industrially viable and reproducible. Particularly, it is desirable to have a process avoiding purification steps still meeting desired pharmaceutical quality.
EXAMPLES
Example-1 : Preparation of isopropyl (Z)-3-(3-(3,5-bis(trifluoromethyl) phenyl)-1 H- -triazol-1 -yl)acrylate
3-(3,5-bis(trifluoromethyl)phenyl)-1 H-1 ,2,4-triazole (250 g) was dissolved in tetrahydrofuran (2 I) under nitrogen atmosphere at 27°C and cooled to -5°C. 1 ,4- diazabicyclo[2.2.2]octane (DABCO, 1 99.5 g) was added to the reaction mixture at -5°C and stirred at the same temperature for 40 minutes. Isopropyl (Z)-3- iodoacrylate (234.8 g in 500 mL of tetrahydrofuran) was added drop wise to the reaction mixture in 1 hour 1 0 minutes at -5°C and stirred at the same temperature for 2 hours. After the completion of the reaction, the reaction mixture was added to ice cold water (2 I) and separated the organic layer. The aqueous layer was extracted with ethyl acetate (2 x 1 I). The combined organic layer was washed with brine solution (1 I) and dried over sodium sulphate. The dried solution was evaporated completely under vacuum at 40°C to obtain crude product with HPLC purity of 93.53% The crude product was triturated with hexane (700 mL) and stirred for 20 minutes at -30°C and filtered the solid. Trituration of crude product with hexane was repeated for three times and dried under vacuum to obtain the title compound with HPLC purity of 97.46% and trans-isomer content of 0.66%. Yield: 297 g Example-2: Preparation of (Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-1 H-1 ,2,4- triazol-1 -yl)acr lic acid.
To a mixture of tetrahydrofuran (300 mL) and water (300 mL), Isopropyl (Z)-3-(3- (3,5-bis(trifluoromethyl)phenyl)-1 H-1 ,2,4-triazol-1 -yl)acrylate (30 g) was added and cooled to 0°C. Lithium hydroxide monohydrate (16.03 g) under cooling condition at 0°C was added to the reaction mixture and stirred the reaction mixture at same temperature for 7 hours. After completion of the reaction, 2 N HCI (180 mL) was added to adjust the pH of the reaction mixture to 2 and extracted it with ethyl acetate (300 mL). Organic layer was dried over sodium sulphate and evaporated under vacuum at 40°C. The crude compound was stirred with hexane (150 mL) and filtered the solid. Dried the compound under vacuum at 40°C for 0.5 hour to obtain the title compound with HPLC purity of 97.25% with trans-isomer content of 3 %. Yield: 24 g
Example-3: Purification of (Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-1 H-1 ,2,4- tria
A mixture of (Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-1 H-1 ,2,4-triazol-1 -yl)acrylic acid (24 g) and acetone (240 mL) was stirred for complete dissolution at 30°C. Dicyclohexyl amine (1 5 mL) was added drop wise for 20 minutes under stirring at the same temperature. Acetone (50 mL) was added to the reaction mixture and stirred for 2 hours at 27°C. Filtered the solid and washed with hot acetone (150 mL) and dried in vacuum drier at 30°C for 1 hour to obtain the Dicyclohexyl amine salt of (Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-1 H-1 ,2,4-triazol-1 -yl)acrylic acid. To the above salt, dichloromethane (150 mL) and water (1 00 mL) was added and stirred for complete dissolution at 30and adjusted the pH of the solution with 2 N sulphuric acid (100 mL) to 2. Filtered the reaction mixture and washed the product with water (1 00 mL) and then with hexane (150 mL). The solid was dried under vacuum at 40°C for 0.5 hour to obtain title compound with HPLC purity 99.98% with no detectable content of trans-isomer. Yield: 17 g
Example-4: Preparation of Selinexor
(Z)-3-(3-(3,5-bis(trifluoromethyl)phenyl)-1 H-1 ,2,4-triazol-1 -yl)acrylic acid (10 g) was combined with a mixture of acetonitrile (1 00 mL) and ethyl acetate (50 mL) then added the 2-hydrazinylpyrazine (3.76 g) and stirred for 5 min. Reaction mixture was cooled to 0°C and diisopropyl ethyl amine (16.63 ml) and then Propylphosphonic anhydride (T3P, 33.31 mL) was added at 0°C and stirred the reaction mixture for 2.5 hours at the same temperature. After completion of the reaction, the reaction mixture was quenched with cold water (100 mL) and extracted the product with ethyl acetate (2 x 150 mL). The combined organic layer was dried over sodium sulphate and evaporated the solvent under vacuum at 40°C to obtain the crude product as yellow syrup. The obtained crude product was combined with dichloromethane (1 00 mL) and filtered the solid and washed with dichloromethane (2 x 50 mL). The solid was dried under vacuum at 40°C to obtain the title compound with purity by HPLC of 99.86%. Yield : 7 g
^ Jump up to:abChen C, Siegel D, Gutierrez M, Jacoby M, Hofmeister CC, Gabrail N (2018). “Safety and efficacy of selinexor in relapsed or refractory multiple myeloma and Waldenstrom macroglobulinemia”. Blood. 131 (8): 855–863. doi:10.1182/blood-2017-08-797886. PMID29203585.
1: Wang AY, Weiner H, Green M, Chang H, Fulton N, Larson RA, Odenike O, Artz AS, Bishop MR, Godley LA, Thirman MJ, Kosuri S, Churpek JE, Curran E, Pettit K, Stock W, Liu H. A phase I study of selinexor in combination with high-dose cytarabine and mitoxantrone for remission induction in patients with acute myeloid leukemia. J Hematol Oncol. 2018 Jan 5;11(1):4. doi: 10.1186/s13045-017-0550-8. PubMed PMID: 29304833.
2: Crochiere ML, Hannus S, Hansen K, Becker F, Baloglu E, Lee M, Kauffman M, Shacham S, Landesman Y. XPO1 target occupancy measurements confirm the selinexor recommended phase 2 dose. Oncotarget. 2017 Nov 30;8(66):110503-110516. doi: 10.18632/oncotarget.22801. eCollection 2017 Dec 15. PubMed PMID: 29299164; PubMed Central PMCID: PMC5746399.
3: Chim CS, Kumar SK, Orlowski RZ, Cook G, Richardson PG, Gertz MA, Giralt S, Mateos MV, Leleu X, Anderson KC. Management of relapsed and refractory multiple myeloma: novel agents, antibodies, immunotherapies and beyond. Leukemia. 2017 Jan 16. doi: 10.1038/leu.2017.329. [Epub ahead of print] Review. PubMed PMID: 29257139.
4: Bobillo S, Abrisqueta P, Carpio C, Raheja P, Castellví J, Crespo M, Bosch F. Promising activity of selinexor in the treatment of a patient with refractory diffuse large B-cell lymphoma and central nervous system involvement. Haematologica. 2017 Dec 14. pii: haematol.2017.181636. doi: 10.3324/haematol.2017.181636. [Epub ahead of print] PubMed PMID: 29242296.
5: Chen C, Siegel D, Gutierrez M, Jacoby M, Hofmeister CC, Gabrail N, Baz R, Mau-Sorensen M, Berdeja JG, Savona M, Savoie L, Trudel S, Areethamsirikul N, Unger TJ, Rashal T, Hanke T, Kauffman M, Shacham S, Reece D. Safety and efficacy of selinexor in relapsed or refractory multiple myeloma and Waldenstrom’s macroglobulinemia. Blood. 2017 Dec 4. pii: blood-2017-08-797886. doi: 10.1182/blood-2017-08-797886. [Epub ahead of print] PubMed PMID: 29203585.
6: Corno C, Stucchi S, De Cesare M, Carenini N, Stamatakos S, Ciusani E, Minoli L, Scanziani E, Argueta C, Landesman Y, Zaffaroni N, Gatti L, Perego P. FoxO-1 contributes to the efficacy of the combination of the XPO1 inhibitor selinexor and cisplatin in ovarian carcinoma preclinical models. Biochem Pharmacol. 2018 Jan;147:93-103. doi: 10.1016/j.bcp.2017.11.009. Epub 2017 Nov 16. PubMed PMID: 29155058.
7: Azmi AS, Li Y, Muqbil I, Aboukameel A, Senapedis W, Baloglu E, Landesman Y, Shacham S, Kauffman MG, Philip PA, Mohammad RM. Exportin 1 (XPO1) inhibition leads to restoration of tumor suppressor miR-145 and consequent suppression of pancreatic cancer cell proliferation and migration. Oncotarget. 2017 Jul 17;8(47):82144-82155. doi: 10.18632/oncotarget.19285. eCollection 2017 Oct 10. PubMed PMID: 29137251; PubMed Central PMCID: PMC5669877.
8: Chen Y, Zhang L, Huang J, Hong X, Zhao J, Wang Z, Zhang K. Dasatinib and chemotherapy in a patient with early T-cell precursor acute lymphoblastic leukemia and NUP214-ABL1 fusion: A case report. Exp Ther Med. 2017 Nov;14(5):3979-3984. doi: 10.3892/etm.2017.5046. Epub 2017 Aug 28. PubMed PMID: 29067094; PubMed Central PMCID: PMC5647690.
9: Body S, Esteve-Arenys A, Miloudi H, Recasens-Zorzo C, Tchakarska G, Moros A, Bustany S, Vidal-Crespo A, Rodriguez V, Lavigne R, Com E, Casanova I, Mangues R, Weigert O, Sanjuan-Pla A, Menéndez P, Marcq B, Picquenot JM, Pérez-Galán P, Jardin F, Roué G, Sola B. Cytoplasmic cyclin D1 controls the migration and invasiveness of mantle lymphoma cells. Sci Rep. 2017 Oct 24;7(1):13946. doi: 10.1038/s41598-017-14222-1. PubMed PMID: 29066743; PubMed Central PMCID: PMC5654982.
10: Broccoli A, Argnani L, Zinzani PL. Peripheral T-cell lymphomas: Focusing on novel agents in relapsed and refractory disease. Cancer Treat Rev. 2017 Nov;60:120-129. doi: 10.1016/j.ctrv.2017.09.002. Epub 2017 Sep 18. Review. PubMed PMID: 28946015.
11: Soung YH, Kashyap T, Nguyen T, Yadav G, Chang H, Landesman Y, Chung J. Selective Inhibitors of Nuclear Export (SINE) compounds block proliferation and migration of triple negative breast cancer cells by restoring expression of ARRDC3. Oncotarget. 2017 May 18;8(32):52935-52947. doi: 10.18632/oncotarget.17987. eCollection 2017 Aug 8. PubMed PMID: 28881784; PubMed Central PMCID: PMC5581083.
12: Garg M, Kanojia D, Mayakonda A, Ganesan TS, Sadhanandhan B, Suresh S, S S, Nagare RP, Said JW, Doan NB, Ding LW, Baloglu E, Shacham S, Kauffman M, Koeffler HP. Selinexor (KPT-330) has antitumor activity against anaplastic thyroid carcinoma in vitro and in vivo and enhances sensitivity to doxorubicin. Sci Rep. 2017 Aug 29;7(1):9749. doi: 10.1038/s41598-017-10325-x. PubMed PMID: 28852098; PubMed Central PMCID: PMC5575339.
13: Conforti F, Zhang X, Rao G, De Pas T, Yonemori Y, Rodriguez JA, McCutcheon JN, Rahhal R, Alberobello AT, Wang Y, Zhang YW, Guha U, Giaccone G. Therapeutic Effects of XPO1 Inhibition in Thymic Epithelial Tumors. Cancer Res. 2017 Oct 15;77(20):5614-5627. doi: 10.1158/0008-5472.CAN-17-1323. Epub 2017 Aug 17. PubMed PMID: 28819023.
14: Arango NP, Yuca E, Zhao M, Evans KW, Scott S, Kim C, Gonzalez-Angulo AM, Janku F, Ueno NT, Tripathy D, Akcakanat A, Naing A, Meric-Bernstam F. Selinexor (KPT-330) demonstrates anti-tumor efficacy in preclinical models of triple-negative breast cancer. Breast Cancer Res. 2017 Aug 15;19(1):93. doi: 10.1186/s13058-017-0878-6. PubMed PMID: 28810913; PubMed Central PMCID: PMC5557476.
15: Schaffer M, Chaturvedi S, Davis C, Aquino R, Stepanchick E, Versele M, Liu Y, Yang J, Lu R, Balasubramanian S. Identification of potential ibrutinib combinations in hematological malignancies using a combination high-throughput screen. Leuk Lymphoma. 2017 Jul 28:1-10. doi: 10.1080/10428194.2017.1349899. [Epub ahead of print] PubMed PMID: 28750570.
16: Muz B, Azab F, de la Puente P, Landesman Y, Azab AK. Selinexor Overcomes Hypoxia-Induced Drug Resistance in Multiple Myeloma. Transl Oncol. 2017 Aug;10(4):632-640. doi: 10.1016/j.tranon.2017.04.010. Epub 2017 Jun 29. PubMed PMID: 28668761; PubMed Central PMCID: PMC5496204.
17: Gupta A, Saltarski JM, White MA, Scaglioni PP, Gerber DE. Therapeutic Targeting of Nuclear Export Inhibition in Lung Cancer. J Thorac Oncol. 2017 Sep;12(9):1446-1450. doi: 10.1016/j.jtho.2017.06.013. Epub 2017 Jun 21. PubMed PMID: 28647672; PubMed Central PMCID: PMC5572747.
18: Machlus KR, Wu SK, Vijey P, Soussou TS, Liu ZJ, Shacham E, Unger TJ, Kashyap T, Klebanov B, Sola-Visner M, Crochiere M, Italiano JE Jr, Landesman Y. Selinexor-induced thrombocytopenia results from inhibition of thrombopoietin signaling in early megakaryopoiesis. Blood. 2017 Aug 31;130(9):1132-1143. doi: 10.1182/blood-2016-11-752840. Epub 2017 Jun 19. PubMed PMID: 28630120; PubMed Central PMCID: PMC5580272.
19: Podar K, Pecherstorfer M. Current and developing synthetic pharmacotherapy for treating relapsed/refractory multiple myeloma. Expert Opin Pharmacother. 2017 Aug;18(11):1061-1079. doi: 10.1080/14656566.2017.1340942. Epub 2017 Jul 5. Review. PubMed PMID: 28604120.
20: Tandon N, Kumar SK. Highlights of Multiple Myeloma at the Annual Meeting of American Society of Hematology, 2016. Indian J Hematol Blood Transfus. 2017 Jun;33(2):153-158. doi: 10.1007/s12288-017-0796-x. Epub 2017 Feb 28. Review. PubMed PMID: 28596644; PubMed Central PMCID: PMC5442069.
Karyopharm’s Selinexor Receives Fast Track Designation from FDA for the Treatment of Patients with Penta-Refractory Multiple Myeloma
NEWTON, Mass., April 10, 2018 (GLOBE NEWSWIRE) — Karyopharm Therapeutics Inc. (Nasdaq:KPTI), a clinical-stage pharmaceutical company, today announced that the U.S. Food and Drug Administration (FDA) has granted Fast Track designation to the Company’s lead, oral Selective Inhibitor of Nuclear Export (SINE) compound selinexor for the treatment of patients with multiple myeloma who have received at least three prior lines of therapy. The FDA’s statement, consistent with the design of Karyopharm’s Phase 2b STORM study, noted that the three prior lines of therapy include regimens comprised of an alkylating agent, a glucocorticoid, Velcade® (bortezomib), Kyprolis® (carfilzomib), Revlimid® (lenalidomide), Pomalyst® (pomalidomide) and Darzalex® (daratumumab). In addition, the patient’s disease must be refractory to at least one proteasome inhibitor (Velcade or Kyprolis), one immunomodulatory agent (Revlimid or Pomalyst), glucocorticoids and to Darzalex, as well as to the most recent therapy. The Company expects to report top-line data from the STORM study at the end of April 2018.
The FDA’s Fast Track program facilitates the development of drugs intended to treat serious conditions and that have the potential to address unmet medical needs. A drug program with Fast Track status is afforded greater access to the FDA for the purpose of expediting the drug’s development, review and potential approval. In addition, the Fast Track program allows for eligibility for Accelerated Approval and Priority Review, if relevant criteria are met, as well as for Rolling Review, which means that a drug company can submit completed sections of its New Drug Application (NDA) for review by FDA, rather than waiting until every section of the NDA is completed before the entire application can be submitted for review.
“The designation of Fast Track for selinexor represents important recognition by the FDA of the potential of this anti-cancer agent to address the significant unmet need in the treatment of patients with penta-refractory myeloma that has continued to progress despite available therapies,” said Sharon Shacham, PhD, MBA, Founder, President and Chief Scientific Officer of Karyopharm. “We are fully committed to working closely with the FDA as we continue development of this potential new, orally-administered treatment for patients who currently have no other treatment options of proven benefit.”
About the Phase 2b STORM Study
In the multi-center, single-arm Phase 2b STORM (Selinexor Treatment of Refractory Myeloma) study, approximately 122 patients with heavily pretreated, penta-refractory myeloma receive 80mg oral selinexor twice weekly in combination with 20mg low-dose dexamethasone, also dosed orally twice weekly. Patients with penta-refractory disease are those who have previously received an alkylating agent, a glucocorticoid, two immunomodulatory drugs (IMiDs) (Revlimid® (lenalidomide) and Pomalyst® (pomalidomide)), two proteasome inhibitors (PIs) (Velcade® (bortezomib) and Kyprolis® (carfilzomib)), and the anti-CD38 monoclonal antibody Darzalex® (daratumumab), and their disease is refractory to at least one PI, at least one IMiD, Darzalex, glucocorticoids and their most recent anti-myeloma therapy. Overall response rate is the primary endpoint of the study, with duration of response and clinical benefit rate being secondary endpoints. All responses will be adjudicated by an Independent Review Committee (IRC).
About Selinexor
Selinexor (KPT-330) is a first-in-class, oral Selective Inhibitor of Nuclear Export (SINE) compound. Selinexor functions by binding with and inhibiting the nuclear export protein XPO1 (also called CRM1), leading to the accumulation of tumor suppressor proteins in the cell nucleus. This reinitiates and amplifies their tumor suppressor function and is believed to lead to the selective induction of apoptosis in cancer cells, while largely sparing normal cells. To date, over 2,300 patients have been treated with selinexor, and it is currently being evaluated in several mid- and later-phase clinical trials across multiple cancer indications, including in multiple myeloma in a pivotal, randomized Phase 3 study in combination with Velcade® (bortezomib) and low-dose dexamethasone (BOSTON), in combination with low-dose dexamethasone (STORM) and as a potential backbone therapy in combination with approved therapies (STOMP), and in diffuse large B-cell lymphoma (SADAL), and liposarcoma (SEAL), among others. Additional Phase 1, Phase 2 and Phase 3 studies are ongoing or currently planned, including multiple studies in combination with one or more approved therapies in a variety of tumor types to further inform Karyopharm’s clinical development priorities for selinexor. Additional clinical trial information for selinexor is available at www.clinicaltrials.gov.
About Karyopharm Therapeutics
Karyopharm Therapeutics Inc. (Nasdaq:KPTI) is a clinical-stage pharmaceutical company focused on the discovery, development and subsequent commercialization of novel first-in-class drugs directed against nuclear transport and related targets for the treatment of cancer and other major diseases. Karyopharm’s SINE compounds function by binding with and inhibiting the nuclear export protein XPO1 (or CRM1). In addition to single-agent and combination activity against a variety of human cancers, SINE compounds have also shown biological activity in models of neurodegeneration, inflammation, autoimmune disease, certain viruses and wound-healing. Karyopharm, which was founded by Dr. Sharon Shacham, currently has several investigational programs in clinical or preclinical development.
/////////Selinexor, FDA 2019, セリネクソル ,KPT-330, KPT 330 , KPT330, AML, Glioma, Sarcoma, Leukemia, Fast Track, CANCER
Phase 1 Clinical, Acute myelogenous leukemia, Protein cereblon modulator
Useful for treating chronic lymphocytic leukemia, chronic myelocytic leukemia, acute lymphoblastic leukemia or acute myeloid leukemia.
Celgene is developing CC-90009, a cereblon E3 ligase modulator, for treating AML; in January 2019, data from a phase I trial were expected later that year.
0iginator Celgene Corporation
Class Antineoplastics
Mechanism of Action CRBN protein modulators; Ubiquitin protein ligase complex modulators
Phase I Acute myeloid leukaemia
28 Mar 2019 No recent reports of development identified for clinical-Phase-Unknown development in Acute-myeloid-leukaemia in USA (IV)
01 Sep 2016 Phase-I clinical trials in Acute myeloid leukaemia (Second-line therapy or greater) in Canada (IV) (NCT02848001)
04 Aug 2016 Celgene plans a phase I trial for Acute Myeloid Leukaemia in USA and Canada (NCT02848001)
In September 2016, Celgene initiated a phase I dose-finding trial of CC 90009 in patients with relapsed or refractory acute myeloid leukaemia (NCT02848001; CC-90009-AML-001). The open-label study intends to enrol 60 patients in the US and Canada
CC-90009 is a cereblon modulator. CC-90009 specifically binds to CRBN, thereby affecting the activity of the ubiquitin E3 ligase complex. This leads to the ubiquitination of certain substrate proteins and induces the proteasome-mediated degradation of certain transcription factors, including Ikaros (IKZF1) and Aiolos (IKZF3), which are transcriptional repressors in T-cells. This reduces the levels of these transcription factors, and modulates the activity of the immune system, which may include the activation of T-lymphocytes. .
Development Overview
cereblon modulator CC-90009A modulator of cereblon (CRBN), which is part of the cullin 4-RING E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase; CUL4-CRBN E3 ubiquitin ligase), with potential immunomodulating and pro-apoptotic activities. Upon administration, CC-90009 specifically binds to CRBN, thereby affecting the activity of the ubiquitin E3 ligase complex. This leads to the ubiquitination of certain substrate proteins and induces the proteasome-mediated degradation of certain transcription factors, including Ikaros (IKZF1) and Aiolos (IKZF3), which are transcriptional repressors in T-cells. This reduces the levels of these transcription factors, and modulates the activity of the immune system, which may include the activation of T-lymphocytes. In addition, this downregulates the expression of other proteins, including interferon regulatory factor 4 (IRF4) and c-myc, which plays a key role in the proliferation of certain cancer cell types. CRBN, the substrate recognition component of the E3 ubiquitin ligase complex, plays a key role in the ubiquitination of certain proteins. Check for active clinical trials using this agent. (NCI Thesaurus)
Provided herein are methods of treating, preventing, managing, and/or ameliorating a hematologic malignancy with 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide or a stereoisomer or a mixture of
stereoisomers, an isotopologue, pharmaceutically acceptable salt, tautomer, solvate, hydrate, co-crystal, clathrate, or polymorph thereof. Further provided is a compound for use in methods of treating, preventing, managing, and/or ameliorating a hematologic malignancy, wherein the compound is 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide or a stereoisomer or a mixture of stereoisomers, an isotopologue, pharmaceutically acceptable salt, tautomer, solvate, hydrate, co-crystal, clathrate, or polymorph thereof.
The term Compound 1 refers to”2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide” having the structure:
and its stereoisomers or mixture of stereoisomers, isotopologues, pharmaceutically acceptable salts, tautomers, solvates, hydrates, co-crystals, clathrates, or polymorphs thereof. In certain embodiments, Compound 1 refers to 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide and its tautomers. In certain embodiments, Compound 1 refers to a polymorph of 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-
oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide. In certain embodiments, Compound 1 refers to polymorph Form C of 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide. In one embodiment, the stereoisomer is an enantiomer.
PATENT
WO-2019136016
Novel isotopologs of the compound presumed to be CC-90009 , processes for their preparation and compositions comprising them are claimed.
SOLID FORMS OF 2-(4-CHLOROPHENYL)-N-((2-(2,6-DIOXOPIPERIDIN-3-YL)-1-OXOISOINDOLIN-5-YL)METHYL)-2,2-DIFLUOROACETAMIDE, AND THEIR PHARMACEUTICAL COMPOSITIONS AND USES
An orally available inhibitor of poly(ADP-ribose) polymerase 1 and 2 (PARP-1/2) for treatment of solid tumors (Jiangsu Hengrui Medicine Co. Ltd., Lianyungang, China)
Fluazolepali, developed by Hengrui and Howson, is intended for the treatment of recurrent ovarian cancer, triple-negative breast cancer, advanced gastric cancer and other advanced solid tumors. Currently, the drug has been introduced into China for recurrent ovarian cancer. Clinical stage.
In February 2019, a randomized, double-blind, controlled, multicenter, phase III clinical study (CTR20190294) of flazopril capsule versus placebo for maintenance of recurrent ovarian cancer was initiated in China and was sponsored by Hengrui Medicine.
Jiangsu Hansoh Pharmaceutical , in collaboration with Jiangsu Hengrui Medicine , is developing an oral capsule formulation of fluazolepali (fluzoparib; SHR-3162), a small molecule inhibitor to PARP-1 and PARP-2, for the treatment of solid tumors including epithelial ovarian, fallopian tube or primary peritoneal, breast and gastric cancer.
Phase I Breast cancer; Fallopian tube cancer; Gastric cancer; Peritoneal cancer; Solid tumours
09 Jul 2019 Jiangsu HengRui Medicine initiates a phase I trial in Solid tumors in China (NCT04013048) [14C]-Fluzoparib
01 Jul 2019 Jiangsu HengRui Medicine plans a phase I drug-drug interaction trial (In volunteers) in China (PO) (NCT04011124)
12 Jun 2019 Jiangsu HengRui Medicine completes a phase I trial in Gastric cancer (Combination therapy, Recurrent, Metastatic disease, Second-line therapy or greater, Late-stage disease) in China (PO) (NCT03026881)
Fluzoparib (SHR 3162) is a selective poly [ADP-ribose] polymerase 1 (PARP1) and poly [ADP-ribose] polymerase 2 inhibitor (PARP2), being developed by Jiangsu HengRui Medicine, for the treatment of cancer. PARP enzymes play a vital role in repair of DNA damage and maintaining genomic stability. Fluzoparib inhibits PARP enzymes and induces DNA-double strands breaks, G2/M arrest and apoptosis in homologous recombination repair (HR)-deficient cells. Clinical development for ovarian cancer, breast cancer, fallopian tube cancer, peritoneal cancer, gastric cancer and solid tumours is underway in China and Australia.
An orally available inhibitor of poly (ADP-ribose) polymerase (PARP) types 1 and 2, with potential antineoplastic activity. Upon oral administration, fluzoparib inhibits PARP 1 and 2 activity, which inhibits PARP-mediated repair of damaged DNA via the base excision repair (BER) pathway, enhances the accumulation of DNA strand breaks, promotes genomic instability, and leads to an induction of apoptosis. The PARP family of proteins catalyze post-translational ADP-ribosylation of nuclear proteins, which then transduce signals to recruit other proteins to repair damaged DNA. PARP inhibition may enhance the cytotoxicity of DNA-damaging agents and may reverse tumor cell chemoresistance and radioresistance. Check for active clinical trials using this agent. (NCI Thesaurus)
Process for preparing heterocyclic compounds (presumed to be fluazolepali ) and its intermediates as PARP inhibitors useful for treating cancer.
Example 1
The compound and 5.0kg of 10% palladium on carbon 250g, 80L of methanol was added to the kettle at 0.4MPa, 24h 25 ℃ hydrogenation reaction. The palladium carbon was removed by filtration, the filter cake was washed with methanol, and the filtrate was collected, evaporated to dryness under reduced pressure, and ethyl acetate (20 L) was added to the concentrate, and the mixture was stirred and evaporated, and then cooled to 0° C. ~3, stirring, filtration, filter cake and then adding 20 L of ethyl acetate, pulping at room temperature for 3 to 4 h, filtration, vacuum drying at 45 ° C for 6-8 h to obtain 5.5 kg of compound 3 solid, yield 91.7%, HPLC purity 99.69%.
Example 2
According to the method of Example 19 of CN102686591A, 2 g of the compound 3 and 2.79 g of the compound 4 were charged to obtain 3.6 g of the compound of the formula I in a yield of 87.8%.
Example 3
At room temperature, 2.0 g of compound 2 (prepared according to the method disclosed in WO2009025784) was dissolved in 30 mL of isopropanol, and concentrated sulfuric acid was added dropwise with stirring to adjust the pH to 3, and stirred at room temperature without solid precipitation; the reaction solution was poured into 150 mL of n-hexane. After stirring at room temperature, no solid precipitated, and the sulfate solid of Compound 2 could not be obtained.
Example 4
1. At room temperature, 1.11 g of compound 2 was dissolved in 10 mL of isopropanol, and 15% phosphoric acid/isopropanol solution was added dropwise with stirring to adjust the pH to 3, stirred at room temperature, filtered, and the filter cake was washed with isopropyl alcohol and dried under vacuum. Compound 2 phosphate solid 1.46 g, yield 87.1%, HPLC purity 99.72%.
Example 5
At room temperature, 1.28 g of compound 2 was dissolved in 10 mL of isopropanol, and 20% acetic acid/isopropanol solution was added dropwise with stirring to adjust the pH to 3, and stirred at room temperature without solid precipitation; the reaction solution was poured into 100 mL of n-hexane, and continued. After stirring at room temperature, no solid precipitated, and the acetate solid of Compound 2 could not be obtained.
Example 6
1.05g of compound 2 was dissolved in 10mL of isopropanol at room temperature, and the pH was adjusted to 3 by adding 15% citric acid/isopropanol solution while stirring. At room temperature, no solid precipitated; the reaction solution was poured into 100 mL of n-hexane. After stirring at room temperature, no solid precipitated, and the citrate solid of Compound 2 could not be obtained.
Example 7
1.12 g of compound 2 was dissolved in 10 mL of isopropanol at room temperature, and 0.74 g of maleic acid was added thereto with stirring. The mixture was stirred at room temperature, filtered, and the filter cake was washed with isopropyl alcohol and dried in vacuo to obtain the maleate salt of compound 2. 1.51 g, yield 84.6%.
SP ROT +3.8 ° Conc: 1.032 g/100mL; methanol; Wavlenght: 589.3 nm, Development of an efficient strategy for the synthesis of the ETB receptor antagonist BQ-788 and some related analogues
Peptides (New York, NY, United States) (2005), 26, (8), 1441-1453., https://doi.org/10.1016/j.peptides.2005.03.022
FOR FREE FORM +19.6 °, Conc: 0.998 g/100mL; : N,N-dimethylformamide; 589.3 nm
presumed to be under license from Banyu , was investigating BQ-788, a selective endothelin receptor B (ETRB) antagonist, for treating metastatic melanoma. By December 2009, the drug was in validation.
Also claimed is their use as an ETBR antagonist and for treating cancers, such as brain cancer, pancreas cancer, colon cancer, breast cancer, ovary cancer, prostate cancer, glioblastoma, solid tumor, melanoma and squamous cell carcinoma. Represent a first filing from ENB Therapeutics Inc and the inventors on these deuterated forms of BQ-788. Melcure SarL ,
SYN
By Brosseau, Jean-Philippe et alFrom Peptides (New York, NY, United States), 26(8), 1441-1453; 2005
N-(cw-2,6-Dimethylpiperidinocarbonyl)-y-methylleucylD-l-(methoxycarbonyl)tryptophanyl-D-norleucine Sodium Salt (1, BQ-788). To a solution of 15 (3.5 g, 5.5 mmol) in methanol (50 mL) was slowly added 5% aqueous NaHCOs (300 mL) over a period of 30 min. The solution was stirred until clarity was achieved (30 min, 23 °C). The solution was diluted with water (200 mL), and the resulting solution was passed through a C18 (60 mL) cartridge preequilbrated in water. BQ-788 (1) was eluted with methanol (2 x 50 mL), concentrated under reduced pressure, resuspended in water (50 mL), and lyophilized to quantitatively yield compound 1 as a white powder:
Novel deuterated analogs of a substituted heterocyclic compound, particularly BQ-788 , processes for their preparation and compositions and combinations comprising them are claimed.
By He, John X.; Cody, Wayne L.; Doherty, Annette M., From Journal of Organic Chemistry (1995), 60(25), 8262-6
Journal of medicinal chemistry (1996), 39(12), 2313-30.
References
^Okada, M; Nishikibe, M (Winter 2002). “BQ-788, a selective endothelin ET(B) receptor antagonist”. Cardiovascular drug reviews. 20 (1): 53–66. PMID12070534.
FDA also approves drug for second indication in a type of lung cancer
The U.S. Food and Drug Administration today granted accelerated approval to Rozlytrek (entrectinib), a treatment for adult and adolescent patients whose cancers have the specific genetic defect, NTRK (neurotrophic tyrosine receptor kinase) gene fusion and for whom there are no effective treatments.
“We are in an exciting era of innovation in cancer treatment as we continue to see development in tissue agnostic therapies, which have the potential to transform cancer treatment. We’re seeing continued advances in the use of biomarkers to guide drug development and the more targeted delivery of medicine,” said FDA Acting Commissioner Ned Sharpless, M.D. “Using the FDA’s expedited review pathways, including breakthrough therapy designation and accelerated approval process, we’re supporting this innovation in precision oncology drug development and the evolution of more targeted and effective treatments for cancer patients. We remain committed to encouraging the advancement of more targeted innovations in oncology treatment and across disease types based on our growing understanding of the underlying biology of diseases.”
This is the third time the agency has approved a cancer treatment based on a common biomarker across different types of tumors rather than the location in the body where the tumor originated. The approval marks a new paradigm in the development of cancer drugs that are “tissue agnostic.” It follows the policies that the FDA developed in a guidance document released in 2018. The previous tissue agnostic indications approved by the FDA were pembrolizumab for tumors with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) tumors in 2017 and larotrectinib for NTRK gene fusion tumors in 2018.
“Today’s approval includes an indication for pediatric patients, 12 years of age and older, who have NTRK-fusion-positive tumors by relying on efficacy information obtained primarily in adults. The FDA continues to encourage the inclusion of adolescents in clinical trials. Traditionally, clinical development of new cancer drugs in pediatric populations is not started until development is well underway in adults, and often not until after approval of an adult indication,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Efficacy in adolescents was derived from adult data and safety was demonstrated in 30 pediatric patients.”
The ability of Rozlytrek to shrink tumors was evaluated in four clinical trials studying 54 adults with NTRK fusion-positive tumors. The proportion of patients with substantial tumor shrinkage (overall response rate) was 57%, with 7.4% of patients having complete disappearance of the tumor. Among the 31 patients with tumor shrinkage, 61% had tumor shrinkage persist for nine months or longer. The most common cancer locations were the lung, salivary gland, breast, thyroid and colon/rectum.
Rozlytrek was also approved today for the treatment of adults with non-small cell lung cancer whose tumors are ROS1-positive (mutation of the ROS1 gene) and has spread to other parts of the body (metastatic). Clinical studies evaluated 51 adults with ROS1-positive lung cancer. The overall response rate was 78%, with 5.9% of patients having complete disappearance of their cancer. Among the 40 patients with tumor shrinkage, 55% had tumor shrinkage persist for 12 months or longer.
Rozlytrek’s common side effects are fatigue, constipation, dysgeusia (distorted sense of taste), edema (swelling), dizziness, diarrhea, nausea, dysesthesia (distorted sense of touch), dyspnea (shortness of breath), myalgia (painful or aching muscles), cognitive impairment (confusion, problems with memory or attention, difficulty speaking, or hallucinations), weight gain, cough, vomiting, fever, arthralgia and vision disorders (blurred vision, sensitivity to light, double vision, worsening of vision, cataracts, or floaters). The most serious side effects of Rozlytrek are congestive heart failure (weakening or damage to the heart muscle), central nervous system effects (cognitive impairment, anxiety, depression including suicidal thinking, dizziness or loss of balance, and change in sleep pattern, including insomnia and excessive sleepiness), skeletal fractures, hepatotoxicity (damage to the liver), hyperuricemia (elevated uric acid), QT prolongation (abnormal heart rhythm) and vision disorders. Health care professionals should inform females of reproductive age and males with a female partner of reproductive potential to use effective contraception during treatment with Rozlytrek. Women who are pregnant or breastfeeding should not take Rozlytrek because it may cause harm to a developing fetus or newborn baby.
Rozlytrek was granted accelerated approval. This approval commits the sponsor to provide additional data to the FDA. Rozlytrek also received Priority Review, Breakthrough Therapy and Orphan Drug designation. The approval of Rozlytrek was granted to Genentech, Inc.
RX518(CK-101) is an orally available third-generation and selective inhibitor of certain epidermal growth factor receptor (EGFR) activating mutations, including the resistance mutation T790M, and the L858R and exon 19 deletion (del 19) mutations, with potential antineoplastic activity.
In August 2019, Suzhou Neupharma and its licensee Checkpoint Therapeutics are developing CK-101 (phase II clinical trial), a novel third-generation, covalent, EGFR inhibitor, as a capsule formulation, for the treatment of cancers including NSCLC and other advanced solid tumors. In September 2017, the FDA granted Orphan Drug designation to this compound, for the treatment of EGFR mutation-positive NSCLC; in January 2018, the capsule was being developed as a class 1 chemical drug in China.
CK-101 (RX-518), a small-molecule inhibitor of epidermal growth factor receptor (EGFR), is in early clinical development at Checkpoint Therapeutics and Suzhou NeuPharma for the potential treatment of EGFR-mutated non-small cell lung cancer (NSCLC) and other advanced solid malignancies.
In 2015, Suzhou NeuPharma granted a global development and commercialization license to its EGFR inhibitor program, excluding certain Asian countries, to Coronado Biosciences (now Fortress Biotech). Subsequently, Coronado assigned the newly acquired program to its subsidiary Checkpoint Therapeutics.
In 2017, the product was granted orphan drug designation in the U.S. for the treatment of EGFR mutation-positive NSCLC.
There are at least 400 enzymes identified as protein kinases. These enzymes catalyze the phosphorylation of target protein substrates. The phosphorylation is usually a transfer reaction of a phosphate group from ATP to the protein substrate. The specific structure in the target substrate to which the phosphate is transferred is a tyrosine, serine or threonine residue. Since these amino acid residues are the target structures for the phosphoryl transfer, these protein kinase enzymes are commonly referred to as tyrosine kinases or serine/threonine kinases.
[0003] The phosphorylation reactions, and counteracting phosphatase reactions, at the tyrosine, serine and threonine residues are involved in countless cellular processes that underlie responses to diverse intracellular signals (typically mediated through cellular receptors), regulation of cellular functions, and activation or deactivation of cellular processes. A cascade of protein kinases often participate in intracellular signal transduction and are necessary for the realization of these cellular processes. Because of their ubiquity in these processes, the protein kinases can be found as an integral part of the plasma membrane or as cytoplasmic enzymes or localized in the nucleus, often as components of enzyme complexes. In many instances, these protein kinases are an essential element of enzyme and structural protein complexes that determine where and when a cellular process occurs within a cell.
[0004] The identification of effective small compounds which specifically inhibit signal transduction and cellular proliferation by modulating the activity of tyrosine and serine/threonine kinases to regulate and modulate abnormal or inappropriate cell proliferation, differentiation, or metabolism is therefore desirable. In particular, the identification of compounds that specifically inhibit the function of a kinase which is essential for processes leading to cancer would be beneficial.
[0005] While such compounds are often initially evaluated for their activity when dissolved in solution, solid state characteristics such as polymorphism are also important. Polymorphic forms of a drug substance, such as a kinase inhibitor, can have different physical properties, including melting point, apparent solubility, dissolution rate, optical and mechanical properties, vapor pressure, and density. These properties can have a direct effect on the ability to process or manufacture a drug substance and the drug product. Moreover, differences in these properties
can and often lead to different pharmacokinetics profiles for different polymorphic forms of a drug. Therefore, polymorphism is often an important factor under regulatory review of the ‘sameness’ of drug products from various manufacturers. For example, polymorphism has been evaluated in many multi-million dollar and even multi-billion dollar drugs, such as warfarin sodium, famotidine, and ranitidine. Polymorphism can affect the quality, safety, and/or efficacy of a drug product, such as a kinase inhibitor. Thus, there still remains a need for polymorphs of kinase inhibitors. The present disclosure addresses this need and provides related advantages as well.
Crystalline form II-VIII of the compound presumed to be CK-101 (first disclosed in WO2015027222 ), for treating a disorder mediated by epidermal growth factor receptor (EGFR) eg cancer.
SCHEME A
Scheme B
General Procedures
Example 1: Preparation of the compound of Formula I (N-(3-(2-((2,3-difluoro-4-(4-(2-hydroxyethyl)piperazin-l-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide)
[0253] To a solution of l,2,3-trifluoro-4-nitrobenzene (2.5 g, 14 mmol, 1.0 eq.) in DMF (20 mL) was added K2C03 (3.8 g, 28 mmol, 2.0 eq.) followed by 2-(piperazin-l-yl)ethanol (1.8 g, 14 mmol, 1.0 eq.) at 0 °C and the mixture was stirred at r.t. overnight. The mixture was poured into ice-water (200 mL), filtered and dried in vacuo to afford 2-(4-(2,3-difluoro-4-nitrophenyl)piperazin-l-yl)ethanol (2.7 g, 67.5%).
[0254] To a solution of 2-(4-(2,3-difluoro-4-nitrophenyl)piperazin-l-yl)ethanol (2.7 g, 9.0 mmol) in MeOH (30 mL) was added Pd/C (270 mg) and the resulting mixture was stirred at r.t.
overnight. The Pd/C was removed by filtration and the filtrate was concentrated to afford 2-(4-(4-amino-2,3-difluorophenyl)piperazin-l-yl)ethanol (2.39 g, 99% yield) as off-white solid.
[0255] To a solution of 8-bromo-2-chloroquinazoline (15.4 g, 63.6 mmol, 1 eq. ) and (3-aminophenyl)boronic acid (8.7 g, 63.6 mmol, 1 eq.) in dioxane/H20 (200 mL/20 mL) was added Na2C03 (13.5 g, 127.2 mmol, 2 eq.), followed by Pd(dppf)Cl2 (2.6 g, 3.2 mmol, 0.05 eq.) under N2, then the mixture was stirred at 80 °C for 12 h. Then the solution was cooled to r.t.,
concentrated and the residue was purified via column chromatography (PE/EA=3 :2, v/v) to afford 3-(2-chloroquinazolin-8-yl)aniline as yellow solid (8.7 g, 53.7% yield).
[0256] To a solution of 3-(2-chloroquinazolin-8-yl)aniline (8.7 g, 34 mmol, 1 eq.) in DCM ( 200 mL ) cooled in ice-bath was added TEA (9.5 mL, 68 mmol, 2 eq. ), followed by acryloyl chloride (4.1 mL, 51 mmol, 1.5 eq.) dropwise. The resulting mixture was stirred at r.t. for 1 h, then washed with brine, dried over anhydrous N2S04 concentrated and the residue was purified via column chromatography (PE/EA=l : 1, v:v) to afford N-(3-(2-chloroquinazolin-8-yl)phenyl)acryl amide as yellow solid(6.6 g, 65% yield).
[0257] To a suspension of 2-(4-(4-amino-2,3-difluorophenyl)piperazin-l-yl)ethanol (83 mg,
0.32 mmol, 1 eq.) and N-(3-(2-chloroquinazolin-8-yl)phenyl)acrylamide (100 mg, 0.32 mmol, 1 eq.) in n-BuOH (5 mL) was added TFA (68 mg, 0.64 mmol, 2 eq.) and the resulting mixture was stirred at 90 °C overnight. The mixture was concentrated, diluted with DCM (20 mL) , washed with Na2C03 solution (20 mL), dried over anhydrous Na2S04, concentrated and the residue was purified via column chromatography (MeOH/DCM=l/30, v:v) to afford N-(3-(2-((2,3-difluoro-4-(4-(2-hydroxyethyl)piperazin-l-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide as a yellow solid(l6.3 mg, 9.5% yield). LRMS (M+H+) m/z calculated 531.2, found 531.2. 1H NMR
Example 2. Preparation of Form I of the compound of Formula I
[0258] Crude compound of Formula I (~30 g, 75% of weight based assay) was dissolved in ethyl acetate (3 L) at 55-65 °C under nitrogen. The resulting solution was filtered via silica gel pad and washed with ethyl acetate (3 L><2) at 55-65 °C. The filtrate was concentrated via vacuum at 30-40 °C to ~2.4 L. The mixture was heated up to 75-85 °C and maintained about 1 hour.
Then cooled down to 50-60 °C and maintained about 2 hours. The heat-cooling operation was repeated again and the mixture was then cooled down to 20-30 °C and stirred for 3 hours. The resulting mixture was filtered and washed with ethyl acetate (60 mL><2). The wet cake was dried via vacuum at 30-40 °C to get (about 16 g) of the purified Form I of the compound of Formula I.
Example 3. Preparation of Form III of the compound of Formula I
[0259] The compound of Formula I (2 g) was dissolved in EtOH (40 mL) at 75-85 °C under nitrogen. n-Heptane (40 mL) was added dropwise into reaction at 75-85 °C. The mixture was stirred at 75-85 °C for 1 hour. Then cooled down to 50-60 °C and maintained about 2 hours. The heat-cooling operation was repeated again and continued to cool the mixture down to 20-30 °C and stirred for 3 hours. The resulting mixture was filtered and washed with EtOH/n-Heptane (1/1, 5 mL><2). The wet cake was dried via vacuum at 30-40 °C to get the purified Form III of the compound of Formula I (1.7 g).
Example 4. Preparation of Form IV of the compound of Formula I The crude compound of Formula I (15 g) was dissolved in ethyl acetate (600 mL) at 75-85 °C under nitrogen and treated with anhydrous Na2S04, activated carbon, silica metal scavenger for 1 hour. The resulting mixture was filtered via neutral Al203 and washed with ethyl acetate (300 mL><2) at 75-85 °C. The filtrate was concentrated under vacuum at 30-40 °C and swapped with DCM (150 mL). n-Heptane (75 mL) was added into this DCM solution at 35-45 °C, and then the mixture was cooled down to 20-30 °C slowly. The resulting mixture was filtered and washed with DCM/n-Heptane (2/1, 10 mL><3). The wet cake was dried via vacuum at 35-40 °C to get the purified Form IV of the compound of Formula I (9.6 g).
Example 5. Preparation of Form V of the compound of Formula I
[0260] Polymorph Form III of the compound of Formula I was dried in oven at 80 °C for 2 days to obtain the polymorph Form V.
Example 6. Preparation of Form VI of the compound of Formula I
[0261] The compound of Formula I (1 g) was dissolved in IPA (20 mL) at 75-85 °C under nitrogen. n-Heptane (20 mL) was added dropwise into reaction at 75-85 °C. The mixture was stirred at 45-55 °C for 16 hours. Then heated up to 75-85 °C and maintained about 0.5 hour.
Then cooled down to 45-55 °C for 0.5 hour and continued to cool the mixture down to 20-30 °C and stirred for 3 hours. Filtered and washed with IPA/n-Heptane (1/1, 3 mL><2). The wet cake was dried via vacuum at 75-80 °C for 2 hours to get the purified Form VI of the compound of Formula I.
Example 7. Preparation of Form VIII of the compound of Formula I
[0262] The polymorph Form VI of the compound of Formula I was dried in oven at 80 °C for 2 days to obtain the polymorph Form VIII.
Example 8. X-ray powder diffraction (XRD)
[0263] X-ray powder diffraction (XRD) patterns were obtained on a Bruker D8 Advance. A CuK source (=1.54056 angstrom) operating minimally at 40 kV and 40 mA scans each sample between 4 and 40 degrees 2-theta. The step size is 0.05°C and scan speed is 0.5 second per step.
Example 9. Thermogravimetric Analyses (TGA)
[0264] Thermogravimetric analyses were carried out on a TA Instrument TGA unit (Model TGA 500). Samples were heated in platinum pans from ambient to 300 °C at 10 °C/min with a nitrogen purge of 60mL/min (sample purge) and 40mL/min (balance purge). The TGA temperature was calibrated with nickel standard, MP=354.4 °C. The weight calibration was performed with manufacturer-supplied standards and verified against sodium citrate dihydrate desolvation.
Example 10. Differential scanning calorimetry (DSC)
[0265] Differential scanning calorimetry analyses were carried out on a TA Instrument DSC unit (Model DSC 1000 or 2000). Samples were heated in non-hermetic aluminum pans from ambient to 300 °C at 10 °C/min with a nitrogen purge of 50mL/min. The DSC temperature was calibrated with indium standard, onset of l56-l58°C, enthalpy of 25-29J/g.
Example 11. Hygroscopicity (DVS)
[0266] The moisture sorption profile was generated at 25°C using a DVS Moisture Balance Flow System (Model Advantage) with the following conditions: sample size approximately 5 to 10 mg, drying 25°C for 60 minutes, adsorption range 0% to 95% RH, desorption range 95% to 0% RH, and step interval 5%. The equilibrium criterion was <0.01% weight change in 5 minutes for a maximum of 120 minutes.
Example 12: Microscopy
[0267] Microscopy was performed using a Leica DMLP polarized light microscope equipped with 2.5X, 10X and 20X objectives and a digital camera to capture images showing particle shape, size, and crystallinity. Crossed polars were used to show birefringence and crystal habit for the samples dispersed in immersion oil.
Example 13: HPLC
[0256] HPLCs were preformed using the following instrument and/or conditions.
///////////////CK-101 , CK 101 , CK101 , phase II , Suzhou Neupharma, Checkpoint Therapeutics , Orphan Drug designation, EGFR mutation-positive NSCLC, NSCLC, CANCER, SOLID TUMOUR, China, RX-518, AK543910
Zanubrutinib, sold under the brand name Brukinsa, is for the treatment of adult patients with mantle cell lymphoma (MCL) who have received at least one prior therapy.[3]
It was approved for medical use in the United States in November 2019.[4][3][5][6]
Zanubrutinib is classified as a Bruton’s tyrosine kinase (BTK) inhibitor. It is administered orally.
History
Efficacy was evaluated in BGB-3111-206 (NCT03206970), a phase II open-label, multicenter, single-arm trial of 86 patients with mantle cell lymphoma (MCL) who received at least one prior therapy.[5] Zanubrutinib was given orally at 160 mg twice daily until disease progression or unacceptable toxicity.[5] Efficacy was also assessed in BGB-3111-AU-003 (NCT 02343120), a phase I/II, open-label, dose-escalation, global, multicenter, single-arm trial of B‑cell malignancies, including 32 previously treated MCL patients treated with zanubrutinib administered orally at 160 mg twice daily or 320 mg once daily.[5][6]
The primary efficacy outcome measure in both trials was overall response rate (ORR), as assessed by an independent review committee.[5] In trial BGB-3111-206, FDG-PET scans were required and the ORR was 84% (95% CI: 74, 91), with a complete response rate of 59% (95% CI 48, 70) and a median response duration of 19.5 months (95% CI: 16.6, not estimable).[5] In trial BGB-3111-AU-003, FDG-PET scans were not required and the ORR was 84% (95% CI: 67, 95), with a complete response rate of 22% (95% CI: 9, 40) and a median response duration of 18.5 months (95% CI: 12.6, not estimable).[5] Trial 1 was conducted at 13 sites in China, and Trial 2 was conducted at 25 sites in the United States, United Kingdom, Australia, New Zealand, Italy, and South Korea.[6]
Yunhang Guo, Ye Liu, Nan Hu, Desheng Yu, Changyou Zhou, Gongyin Shi, Bo Zhang, Min Wei, Junhua Liu, Lusong Luo, Zhiyu Tang, Huipeng Song, Yin Guo, Xuesong Liu, Dan Su, Shuo Zhang, Xiaomin Song , Xing Zhou, Yuan Hong, Shuaishuai Chen, Zhenzhen Cheng, Steve Young, Qiang Wei, Haisheng Wang, Qiuwen Wang, Lei Lv, Fan Wang, Haipeng Xu, Hanzi Sun, Haimei Xing, Na Li, Wei Zhang, Zhongbo Wang, Guodong Liu, Zhijian Sun, Dongping Zhou, Wei Li, Libin Liu, Lai Wang, Zhiwei Wang
Aberrant activation of Bruton’s tyrosine kinase (BTK) plays an important role in pathogenesis of B-cell lymphomas, suggesting that inhibition of BTK is useful in the treatment of hematological malignancies. The discovery of a more selective on-target covalent BTK inhibitor is of high value. Herein, we disclose the discovery and preclinical characterization of a potent, selective, and irreversible BTK inhibitor as our clinical candidate by using in vitro potency, selectivity, pharmacokinetics (PK), and in vivo pharmacodynamic for prioritizing compounds. Compound BGB-3111 (31a, Zanubrutinib) demonstrates (i) potent activity against BTK and excellent selectivity over other TEC, EGFR and Src family kinases, (ii) desirable ADME, excellent in vivo pharmacodynamic in mice and efficacy in OCI-LY10 xenograft models.
Bruton’s tyrosine kinase (Btk) belongs to the Tec tyrosine kinase family (Vetrie et al., Nature 361: 226-233, 1993; Bradshaw, Cell Signal. 22: 1175-84, 2010). Btk is primarily expressed in most hematopoietic cells such as B cells, mast cells and macrophages (Smith et al., J. Immunol. 152: 557-565, 1994) and is localized in bone marrow, spleen and lymph node tissue. Btk plays important roles in B-cell receptor (BCR) and FcR signaling pathways, which involve in B-cell development, differentiation (Khan, Immunol. Res. 23: 147, 2001). Btk is activated by upstream Src-family kinases. Once activated, Btk in turn phosphorylates PLC gamma, leading to effects on B-cell function and survival (Humphries et al., J. Biol.Chem. 279: 37651, 2004).
[0003] These signaling pathways must be precisely regulated. Mutations in the gene encoding Btk cause an inherited B-cell specific immunodeficiency disease in humans, known as X-linked agammaglobulinemia (XLA) (Conley et al., Annu. Rev. Immunol. 27: 199-227, 2009). Aberrant BCR-mediated signaling may result in dysregulated B-cell activation leading to a number of autoimmune and inflammatory diseases. Preclinical studies show that Btk deficient mice are resistant to developing collagen- induced arthritis. Moreover, clinical studies of Rituxan, a CD20 antibody to deplete mature B-cells, reveal the key role of B-cells in a number of inflammatory diseases such as rheumatoid arthritis, systemic lupus erythematosus and multiple sclerosis (Gurcan et al, Int. Immunopharmacol. 9: 10-25, 2009). Therefore, Btk inhibitors can be used to treat autoimmune and/or inflammatory diseases.
[0004] In addition, aberrant activation of Btk plays an important role in pathogenesis of B-cell lymphomas indicating that inhibition of Btk is useful in the treatment of hematological malignancies (Davis et al, Nature 463: 88-92, 2010). Preliminary clinical trial results showed that the Btk inhibitor PCI-32765 was effective in treatment of several types of B-cell lymphoma (for example, 54thAmerican Society of Hematology (ASH) annual meeting abstract, Dec. 2012: 686 The Bruton’s Tyrosine Kinase (Btk) Inhibitor, Ibrutinib (PCI- 32765), Has Preferential Activity in the ABC Subtype of Relapsed/Refractory De Novo Diffuse Large B-Cell Lymphoma (DLBCL): Interim Results of a Multic enter, Open-Label, Phase I Study). Because Btk plays a central role as a mediator in multiple signal transduction pathways, inhibitors of Btk are of great interest as anti-inflammatory and/or anti-cancer agents {Mohamed et al., Immunol. Rev. 228: 58-73, 2009; Pan, Drug News perspect 21: 357-362, 200%; Rokosz et al., Expert Opin. Ther. Targets 12: 883-903, 2008; Uckun et al., Anti-cancer Agents Med. Chem. 7: 624-632, 2007; Lou et al, J. Med. Chem. 55(10): 4539-4550, 2012).
[0005] International application WO2014173289A disclosed a series of fused heterocyclic compounds as Btk inhibitors. In particular, WO2014173289A disclosed
(S)-7-(l-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl)-4,5,6,7-tetra-hydropyrazolo[l,5-a]pyrimi dine-3-carboxamide (hereinafter C
Compound 1
[0006] Compound 1 is a potent, specific and irreversible BTK kinase inhibitor. The data generated in preclinical studies using biochemical, cell based and animal studies suggested that Compound 1 could offer significant benefit in inhibiting tumor growth in B-cell malignancies. As Compound 1 was shown to be more selective than ibrutinib for inhibition of BTK vs. EGFR, FGR, FRK, HER2, HER4, ITK, JAK3, LCK, and TEC, it is expected to give rise to less side-effects than ibrutinib in clinic. In addition, Compound 1 showed significantly less inhibition of rituximab-induced antigen-dependent cell-mediated cytotoxicity (ADCC) than ibrutinib due to weaker ITK inhibition, and therefore may provide better efficacy when combined with rituximab or other ADCC-dependent antibody in treating B-cell malignancies.
[0007] Preclinical safety evaluation has demonstrated that Compound 1 was safer than ibrutinib in terms of the overall tolerance and severe toxicities in both rat and dog single and repeat dose toxicity studies up to 28 days. Additionally, Compound 1 had better bioavailability without accumulation issues observed for ibrutinib. These unique characteristics warrant further evaluation of Compound 1 in clinical studies.
[0008] However, Compound 1 was found to be an amorphous form according to the preparation method for Compound 27 in WO 2014173289A, which was further confirmed by the X-Ray Powder Diffraction pattern of FIG. 7A. The amorphous form was shown to have a low glass transition temperature as shown in FIG. 7B, indicating some difficulties in the drug formulation with the amorphous form, such as low stability and hard to purify. Therefore, it’s necessary to develop a new form of Compound 1 which possesses characteristics such as high melting point and better stability, suitable for drug formulation.
Scheme 1: Preparation of Compound 1 and deuterium-labeled Compound 1
[0105] Under N2 atmosphere, ACN (12.0 v), water (12.5 v), BG-13 (8.0 Kg, 1.0 eq), and NaHC03 (2.5 eq.) were added to a reactor. The mixture was then cooled to -5-0 °C. To the mixture, the solution of acryloyl chloride (1.1 eq.) in MeCN (0.5 v) was added dropwise and
stirred until the reaction was completed. EA (6.0 v) was then added to the reactor, and stirred. The organic phase was collected. The aqueous layer was further extracted with EA (3.0 v). The organic phases were combined and washed with brine. The organic layer was collected and concentrated.
[0106] The residue was purified by silica gel (2 wt) column, eluted with 3% w/w methanol in DCM (21.0 v). The Compound 1 solution was collected and concentrated under vacuum. The residue was precipitated from EA/MTBE (2.0 v). The cake was collected by centrifugation as the product.
Step 15: Synthesis of (S)-7-(l-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl)
-4,5,6,7-tetrahydropyrazolori,5-a1pyrimidine-3-carboxamide (Compound 1, alternative method)
[0107] A mixture of CHsCN (10.0 v), purified water (5.0 v), NaOH (1.5 eq.) and BG-13 (1.0 eq.) was stirred to get a clear solution. EtOAc (6.0 v) was then charged to the reaction and separated. The organic phase was collected and washed with 15% brine (3.0 v) twice. The organic phase prepared above was concentrated and the solvent was swapped to CH3CN (residue volume: NMT 5.0 v). CH3CN (7.5 v) and purified water (12.5 v) were charged and cooled to 15-20°C. L-(+)-tartaric acid (0.5 eq) and NaHCCb (2.5 eq.) were charged to the reaction mixture. A solution of acryloyl chloride (1.1 eq.) in CH3CN (0.5 v) was charged drop-wise to the reaction mixture. After the reaction was completed, EtOAc (6.0 v) was charged to the reaction mixture and organic layer was collected. Aqueous phase was further extracted with EA (3.0 v). The organic layers were combined, washed with 15% brine (5.0 v) and concentrated. The solvent was swapped to DCM (volume of residue: 1.5-2.0 v) and purified by silica gel column (silica gel: 100-200 mush, 2.0 w/ w; eluent: 3%> w/ w MeOH in DCM (about 50 v). The collected solution was concentrated and swapped to EtOAc (4.0 v). MTBE (6.4 v) was charged drop-wise to residue at 50°C. The mixture was then cooled to 5°C and the cake was collected centrifugation.
Step 16: Preparation of Crystalline Form A of Compound 1
[0108] The above cake of Compound 1 was dissolved in 7.0 volumes of DCM, and then swapped to solvent EA. After recrystallization from EA/MTBE, the cakes was collected by centrifugation, and was dried under vacuum. This gave 4.44 Kg product (Yield: 70.2%).
[0109] The product was then characterized by X-ray powder diffraction (XRPD) pattern method, which was generated on a PANalytical Empyrean X-ray powder diffractometer with the XRPD parameters as follows: X-Ray wavelength (Cu, ka, Kal (A): 1.540598, Ka2(A): 1.544426; Ka2/Kal intensity ratio: 0.50); X-Ray tube setting (45 Kv, 40mA); divergence slit (automatic); scan mode (Continuous); scan range (°2TH) (3°-40); step size (°2TH) (0.0131); scan speed (°/min) (about 10). The XRPD result found the resultant product as a crystalline shown in FIG. 1.
[0110] The differential scanning calorimetry (DSC) curves shown as in FIG. 2 was generated on a TA Q2000 DSC from TA Instruments. The DSC parameters used includes: temperature (25°C-desired temperature); heating rate (10°C/min) ; method (ramp); sample pan (aluminum, crimped); purge gas (N2). DSC result showed a sharp melting point at 139.4°C (onset temperature).
[0111] The thermo-gravimetric analysis (TGA) curves shown as in FIG. 3 was generated on a TA Q5000 TGA from TA Instruments. The TGA parameters used includes: temperature
(RT-desired temperature); heating rate (10°C/min); method (ramp); sample pan (platinum, open); purge gas (N2). TGA result showed is anhydrous with no weight loss even up to 110 °C.
[0113] The carbon nuclear magnetic resonance (13C-NMR) shown as in FIG. 5 was collected on a Bruker 400M NMR Spectrometer in DMSO-de. 13C-NMR spectra for Crystalline Form A of Compound 1.
Step 15: Synthesis of (S)-7-(1-acrvlovlpiperidin-4-vl)-2-(4-phenoxvphenyl)-4.5.6.7-tetrahvdropvrazolo[1.5-a1pvrimidine-3-carboxamide (Compound 1)
[0119] Under N2 atmosphere, ACN (12.0 v), water (12.5 v), BG-13 (8.0 Kg, 1.0 eq), and NaHCO3 (2.5 eq.) were added to a reactor. The mixture was then cooled to -5-0 °C. To the mixture, the solution of acryloyl chloride (1.1 eq.) in MeCN (0.5 v) was added dropwise and stirred until the reaction was completed. EA (6.0 v) was then added to the reactor, and stirred. The organic phase was collected. The aqueous layer was further extracted with EA (3.0 v). The organic phases were combined and washed with brine. The organic layer was collected and concentrated.
[0120] The residue was purified by silica gel (2 wt) column, eluted with 3% w/w methanol in DCM (21.0 v). The Compound 1 solution was collected and concentrated under vacuum. The residue was precipitated from EA/MTBE (2.0 v). The cake was collected by centrifugation as the product.
Step 15: Synthesis of (S)-7-(l-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl) -4.5.6.7-tetrahvdropvrazolori.5-a1pvrimidine-3-carboxamide (Compound 1. alternative method)
[0121] A mixture of CH3CN (10.0 v), purified water (5.0 v), NaOH (1.5 eq.) and BG-13 (1.0 eq.) was stirred to get a clear solution. EtOAc (6.0 v) was then charged to the reaction and separated. The organic phase was collected and washed with 15% brine (3.0 v) twice. The organic phase prepared above was concentrated and the solvent was swapped to CH3CN (residue volume: NMT 5.0 v). CH3CN (7.5 v) and purified water (12.5 v) were charged and cooled to 15-20°C. L-(+)-tartaric acid (0.5 eq) and NaHCO3 (2.5 eq.) were charged to the reaction mixture. A solution of acryloyl chloride (1.1 eq.) in CH3CN (0.5 v) was charged drop-wise to the reaction mixture. After the reaction was completed, EtOAc (6.0 v) was charged to the reaction mixture and organic layer was collected. Aqueous phase was further extracted with EA (3.0 v). The organic layers were combined, washed with 15% brine (5.0 v) and concentrated. The solvent was swapped to DCM (volume of residue: 1.5-2.0 v) and purified by silica gel column (silica gel: 100-200 mush, 2.0 w/ w; eluent: 3% w/ w MeOH in DCM (about 50 v). The collected solution was concentrated and swapped to EtOAc (4.0 v). MTBE (6.4 v) was charged drop-wise to residue at 50°C. The mixture was then cooled to 5°C and the cake was collected centrifugation.
Celgene (now a wholly owned subsidiary of Bristol-Myers Squibb ) , following its acquisition of Quanticel , is developing CC-90010, an oral inhibitor of BET (bromodomain and extraterminal) proteins, for the potential treatment of solid tumors and non-Hodgkin’s lymphoma. In August 2019, a phase I trial for diffuse astrocytoma, grade III anaplastic astrocytoma and recurrent glioblastoma was planned
PATENT
WO2018075796 claiming solid composition comprising a bromodomain inhibitor, preferably 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-1-one in crystalline form A.
[00344] A suspension of 4-bromo-2-methylisoquinolin-l-one (100 mg, 0.42 mmol), bis(pinacolato)diboron (214 mg, 0.84 mmol), Pd(dppf)Cl2 (31 mg, 0.04 mmol) and potassium acetate (104 mg, 1.05 mmol) in dioxane (2 mL) under nitrogen was warmed up to 90 °C for 135 minutes. It was then cooled down to room temperature and diluted with ethyl acetate (8 mL). The mixture was washed with aqueous saturated solution of NaHC03 (8 mL) and brine (8 mL). The organic phase was separated, dried over Na2S04, filtered and concentrated under reduced pressure. The residue was purifed by normal phase column chromatography (10-90% EtOAc/Hexanes) to give the title compound (44 mg, 37%). 1H NMR (CDC13, 400 MHz) δ 8.43 (d, J = 7.9 Hz, 1 H), 8.40 (dd, J = 8.2 Hz, 0.9 Hz, 1 H), 7.68 (s, 1 H), 7.65 (ddd, J = 8.2, 8.2, 1.1 Hz, 1 H), 7.46 (t, J = 7.5 Hz, 1 H), 3.63 (s, 3H), 1.38 (s, 12H). LCMS (M+H)+ 286. Step 2: 4-[2-(cyclopropylmethox -5-methylsulfonylphenyl]-2-methylisoquinolin-l-one
[00345] The title compound was prepared in a manner similar to Example 18, step 3, substituting 2-bromo-l-(cyclopropylmethoxy)-4-methylsulfonylbenzene for 4-bromo-2-methylisoquinolin-l(2H)-one and 2-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)isoquinolin-l-one for N-benzyl-2-methoxy-5-(tetramethyl-l,3,2-dioxaborolan-2-yl)benzamide. 1H NMR (DMSO-d6, 400 MHz) δ 0.09 (m, 2 H), 0.29 (m, 1H), 0.35 (m, 1H),
[00347] A mixture of 4-bromo-2-methylisoquinolin-l-one (8.0 g, 33.6 mmol),
bis(pinacolato)diboron (17.1 g, 67.2 mmol), KOAc (6.6 g, 67.2 mmol), Pd2(dba)3 (3.1 g, 3.36 mmol) and X-Phos (1.6 g, 3.36 mmol) in anhydrous dioxane (200 mL) was stirred at 60 °C for 12 h. The reaction mixture was concentrated and the residue was purified by column chromatography on silica gel (PE : EA = 15 : 1) to give the title compound (6.0 g, 62 %) as a solid.
[00348] The title compound from Step 1 (5.0 g, 17.5 mmol), 2-bromo-l-(cyclopropylmethoxy)-4-methylsulfonylbenzene (6.4 g, 21 mmol), K3PO4 (9.3 g, 43.9 mmol) and Pd(dppf)Cl2 (1.4 g, 1.75 mmol) in a dioxane/water (100 mL / 10 mL) mixture were stirred at 60 °C for 12 hrs. The reaction mixture was concentrated under reduced pressure and the residue was purified by column chromatography on silica gel (EA : DCM = 1 : 4).
Appropriate fractions were combined and concentrated under reduce pressure. The resultant solid was recrystallized from DCM / MTBE (1 : 1, 50 mL) to give the title compound (4.0 g, 60 %) as a white solid. 1H NMR: (CDC13, 400 MHz) δ 8.51 (dd, Ji = 8.0 Hz, J2 = 0.8 Hz, 1 H), 7.98 (dd, Ji = 8.4 Hz, J2 = 2.4 Hz, 1 H), 7.86 (d, J = 2.4 Hz, 1 H), 7.53 (m, 2 H), 7.16 (d, J = 7.6 Hz, 1 H), 7.10 (m, 2 H), 3.88 (m, 2 H), 3.66 (s, 3 H), 3.09 (s, 3 H), 1.02-0.98 (m, 1 H), 0.44-0.38 (m, 2 H), 0.11-0.09 (m, 2 H). LCMS: 384.1 (M+H)+
A process for preparing bromodomain inhibitor, particularly 4-[2(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-1-one (having HPLC purity of 99%; compound 1; (hereafter referred to as C-90010)) and its hydrates, solvates, prodrugs and salts comprising the reaction of a substituted 4-(methylsulfonyl)phenol compound with a quinoline derivative, followed by purification is claimed. Also claimed are novel intermediates of CC-90010 and their processes for preparation. Further claimed are novel crystalline form of CC-90010. CC-90010 is known and disclosed to be a bromodomain containing protein inhibitor, useful for treating cancer.
Scheme 10: Synthesis of Compound 1
[0090] Acetonitrile (1.6L) was charged to a mixture of Compound 2 (156.7g, 460 mmol), Compound 3 (lOOg, 420 mmol) and potassium phosphate tribasic (223g, l.OSmol). Agitation
was begun and water (400mL) charged to the batch. The system was vacuum purged three times with nitrogen and charged with Pd(PPh3)2Cl2 (2.9g, 4 mmol) and the system vacuum purged three times with nitrogen. The batch was heated to about 65 to about 75 °C (or any temperature in between and including these two values) and contents stirred for at least about 16 hours until reaction was complete by HPLC analysis. The batch was cooled to about 60 to about 70 °C (or any temperature in between and including these two values), agitation halted and the mixture allowed to settle. The bottom aqueous layer was removed. Water (150mL) and acetonitrile (700mL) were charged at about 60 to about 70°C (or any temperature in between and including these two values). Ecosorb C-941 (15g) and Celite (lOg) were charged to the reaction vessel at about 60 to about 70°C (or any temperature in between and including these two values). After lh, the mixture was filtered to remove solids. The solids were washed twice each with 18% water in acetonitrile (500 mL) at about 60 to about 70°C (or any temperature in between and including these two values). The filtrates were combined and concentrated under atmospheric pressure to a final volume of 1.5L. The batch was cooled to about 60 to about 65°C (or any temperature in between and including these two values) and seeded with Compound 1 (1 g). After lh, water (500 mL) was charged over at least 1 hour at about 60 to about 65°C (or any temperature in between and including these two values). The slurry was cooled to about 15 to about 25°C (or any temperature in between and including these two values) over 4 hours. The product was collected by suction filtration. The wet cake was washed with 45% water in acetonitrile (500mL) twice. The product was dried under vacuum at about 40°C with nitrogen purge. Yield: 139g of 1.
[0091] The above procedure for coupling Compound 3 and Compound 2 to produce
Compound 1 may be modified in any of the ways that follow. Reaction solvents: Different reaction solvents from acetonitrile can be used, including tetrahydrofuran, 2-methyl tetrahydrofuran, toluene, and isopropanol. Boronic ester: Different boronic esters from Compound 2 can be used, including pinacolato ester compound 7, and the free boronic acid of Compound 2. Examples of boronic esters can be found in Lennox et al., Chem. Soc. Rev., 43: 412 (2014). Carbon treatment: Different carbon treatments from Ecosorb C-941 could be used. Different amounts of carbon, from 0.01 to 0.5X weight can be used. The carbon can be eliminated. Different amounts of Celite, from 0.01 to 0.5X weight can be used.
Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used.
The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying. Catalysts: Different metal and ligand combination could be used. Examples of metal/ligand combinations can be found in Maluenda, Irene; Navarro, Oscar, Molecules, 2015, 20, 7528. Various catalysts can be including: XPhos-3G (cas# 1445085-55-1); cataCXium® A Pd 3G (CAS# 1651823-59-4); PdCk(DtBPF) (CAS# 95408-45-0); SPhos 3G (Cas# 1445085-82-4); AmPhos 3G (Cas# 1820817-64-8); PCy3 3G (Cas# 1445086-12-3); Pd PEPPSI IPent Cas#l 158652-41-5);
Pd(PPh3)2Cb (Cas# 13965-03-2). Examples of catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
VI. Purification of Compound 1 fCC-900101 bv crystallization from formic acid and water
[0092] Described herein are methods of purifying Compound 1 by crystallization from formic acid and water. Also described are methods for obtaining three different polymorphs of Compound 1, including the most stable form, Form 1 and two metastable forms, Form 4
The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying. Catalysts: Different metal and ligand combination could be used. Examples of metal/ligand combinations can be found in Maluenda, Irene; Navarro, Oscar, Molecules, 2015, 20, 7528. Various catalysts can be including: XPhos-3G (cas# 1445085-55-1); cataCXium® A Pd 3G (CAS# 1651823-59-4); PdCh(DtBPF) (CAS# 95408-45-0); SPhos 3G (Cas# 1445085-82-4); AmPhos 3G (Cas# 1820817-64-8); PCy3 3G (Cas# 1445086-12-3); Pd PEPPSI IPent Cas#l 158652-41-5);
Pd(PPh3)2Cl2 (Cas# 13965-03-2). Examples of catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
VI. Purification of Compound 1 (CC-90010! bv crystallization from formic acid and water
[0092] Described herein are methods of purifying Compound 1 by crystallization from formic acid and water. Also described are methods for obtaining three different polymorphs of Compound 1, including the most stable form, Form 1 and two metastable forms, Form 4
33 -a
and Form 5. Supporting data (XRPD, DSC, photomicroscopy) for all three forms is provided in the examples below.
[0093] The stmcture of Compound 1 (CC-90010) is shown below:
Example 1: Synthesis of Compound 1
[0217] Synthesis of compound 1 was accomplished according to Scheme 1 below. Referring to Scheme 1, synthesis commenced with bromination of starting material 4-(methylsulfonyl)phenol 4, to produce compound 5. Compound 5 was O-alkylated with (bromomethyl)cyclopropane to produce compound 6. Boronate Compound 2 was then formed by borylation of Compound 6 with Pd catalyst and bis(pinacolato)diboron to produce transient Compound 7, which was subsequenctly treated with diethanolamine (DBA) to afford cross-coupling partner Compound 2. Cross-coupling partner Compound 3 was formed in one pot starting from commercially available Compound 8. Compound 8 was N-methylated and brominated to afford Compound 3. Compounds 2 and 3 were cross-coupled (Norio, M. and Suzuki, A., Chem. Rev., 95(7), 2457-2483 (1995)) to afford the target compound 1.
Scheme 1: Synthesis of compound 1
1.1: Bromination of 4
[0218] The bromination of Compound 4 to produce Compound 5 itself is simple, however stopping at the mono-brominated Compound 5 was challenging. The bis-brominated Compound 5-a (see Scheme 2 below) is a particularly pernicious impurity as it couples downstream to form a di ffi cult-to-purge impurity.
Scheme 2: Bromination of Compound 4
[0219] The key to high purity with reasonable yield was to exploit the solubility differences of the starting material Compound 4 (46 mg/ml at 20 °C) and the product Compound 5 (8 mg/ml) in CH2CI2. These solubility differences are summarized in Table 3 below.
[0220] This solubility difference is exploited by performing the reaction at a high
concentration to drive Compound 5 out of solution once formed, thereby minimizing its ability to react further with the brominating reagent to form Compound 5-a diBr. The reaction is seeded with Compound 5 to initiate its crystallization.
[0221] In Fig. 22 (Conversion of Compound 4 to Compound 5: Effect of Sulfuric Acid) it can be seen that in the absence of acid the initial reaction to Compound 5 is rapid, however the conversion plateaus at about 30% Compound 5. The main side product was found to be the impurity Compound 5-a diBr (see Fig. 23: Conversion of Compound 5 and Compound 5-a diBr: No H2SO4). Addition of increasing amounts of sulfuric acid leads to a higher conversion to desired Compound 5.
[0222] Fig. 24 (Compound 4 to Compound 5 Reaction Profile: Portion-wise Addition of NBS, Seeding) depicts further reaction control. The portion-wise addition ofNBS after addition of catalytic sulfuric acid minimizes the temperature rise, and the addition of Compound 5 after an initial NBS charge promotes the reactive crystallization of Compound 5. After about 6 to 7 hours of reaction it can be seen that the major product is Compound 5, with only a small (<5%) of the di-brominated impurity formed. In contrast, in a reaction where Compound 4 and all of the NBS were charged followed by the addition of 4 volumes of methylene chloride, a rapid exotherm resulted and undesired Compound 5-a diBr was found to be the major product.
[0223] Thus, the reaction was run under a high concentration in CH2CI2 with a portion-wise solid addition of NBS (to control both availability of the electrophile and the exotherm). An end of reaction slurry sample typically showed not more than 5% of the starting material Compound 4 remaining. After filtration the crude cake was washed with cold CH2CI2 and the OkCk-washed filter cake contained not more than 0.5% by weight dibrominated Compound 5-a. It also contained a large amount of HPLC-silent succinimide.
[0224] The following procedure was carried out: Compound 4 (25g, 145mmol) followed by CH2CI2 (lOOmL) were added to a reaction vessel and agitated. The batch was adjusted to 17 °C to 23 °C. Sulfuric acid was charged (2.7mL, Slmmol) to the batch maintaining 17 °C to 23°C. The batch was stirred at 17 °C to 23 °C for 10 minutes to 20 minutes. The first portion of A-bromosuccimide (NBS) was charged (6.5g, 36.5 mmol) to the batch at 17 °C to 23°C and stirred for at least 30 min. The second portion of NBS was charged (6.5g, 36.5 mmol) to the batch at 17 °C to 23°C and stirred for at least 30 min. The batch was seeded with
Compound 5 (0.02wt) and stirred for ca. 30 min at 17 °C to 23 °C to induce crystallization.
[0225] The third portion of NBS was charged (6.5g, 36.5 mmol) to the batch at 17 °C to 23 °C and stirred for at least 30 min. NBS (6.5g, 36.5 mmol) was charged to the batch at 17 °C to 23 °C and stirred for at least 30 min. Additional CH2CI2 was charged (50mL) to the batch while maintaining 17 °C to 23 °C to aid in agitation and transfer for filtration. The batch was stirred at 17 °C to 23 °C until complete by HPLC analysis (~20 – 40 h). The product was collected by suction filtration. The filter cake was slurry washed with CH2CI2 (3 x 50mL) at 17 °C to 23 °C (target 20 °C). The filter cake was slurry washed with purified water (3.0vol) at 65 °C to 75 °C for 2 to 3 hours. Then, the filter cake was slurry washed with purified water (3 x 1.0 vol, 3 x 1.0 wt) at 17 °C to 23°C. The wet cake was dried under vacuum with nitrogen bleed at 60 °C. Yield: 27g 5 (74% molar) >97% by weight. ¾ NMR (500 MHz, de-DMSO) 8.01 (1H, d, 4J = 2.1 Hz, RO-Ar meta- H ), 7.76 (1H, dd, J = 8.6 and 4J = 2.1 Hz, RO-Ar meta-H ), 7.14 (1H, d, J = 8.6 Hz, RO-Ar ortho- H), 3.38 (1H, br s, OH), 3.20 (3H, s,
CHJ); MS (ES-) calc. 249/251; found 249/251. Melting point (MP): (DSC) 188 °C.
[0226] The above procedure allowed for the following modifications. Solvents: Alternative solvents could be used. Examples include chlorinated solvents, such as chloroform or 1,2 dichloroethane, and non-chlorinated solvents such as acetonitrile, tetrahydrofuran, or 2- methyltetrahydrofuran. Reaction concentration: The reaction concentration can be varied from about 2X vol to about 20 X vol (with respect to Compound 4). Brominating agents: Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination reagent stoichiometry: Different amounts of the brominating reagent can be used, from about 0.8 equiv to about 1.9 equiv. Bromination reagent addition: The brominating reagent can be added all at once, portion wise in about 2 to about 20 portions, or continuously. The addition times can vary from about 0 to about 72 hours. Temperature: Reaction temperatures from about 0 °C to about 40 °C could be used. Acids: Different acids can be envisioned, including benzenesulfonic acid, para-toluenesulfonic acid, triflic acid, hydrobromic acid, and trifluoroacetic acid. Isolation: Instead of directly filtering the product and washing with methylene chloride and water, at the end of reaction an organic solvent capable of dissolving Compound 5 could be charged, followed by an aqueous workup to remove succinimide, and addition of an antisolvent or solvent exchange to an appropriate solvent to crystallize Compound 4. Drying: A temperature range of about 10 to about 60 °C could be used for drying.
[0227] An alternative process to Compound 5 has also been developed. This process is advantageous in that it does not use a chlorinated solvent, and provides additional controls over the formation of the Compound 5-a dibromo impurity. See Oberhauser, T. J Org. Chem 1997, 62, 4504-4506. The process is as follows. Compound 4 (10 g, 58 mmol) and acetonitrile (100 ml) were charged to the reactor and agitated. The batch was cooled to -20 °C. Triflic acid (CF3SO3H or TfOH, 5.5 mL, 62 mmol) was charged while maintaining a batch temperature of -10 to -25 °C. N-bromosuccinimide was charged (NBS, 11.4 g, 64 mmol), stirred at -10 to -25 °C for 30 minutes, then warmed to 20 °C over 3 to 4 hours. Agitation was continued at 15 °C to 25 °C until reaction completion. If the reaction conversion plateaued before completion, the reaction was cooled to -5 to -15 °C, and additional NBS was added, the amount based off of unreacted starting material, followed by warming to 15 °C to 25 °C and reacting until complete.
[0228] After reaction completion, the batch was warmed to 40 °C to 50 °C and concentrated under reduced pressure to 40 mL. The batch was cooled to -5 °C to -15 °C and the resulting product solids were filtered off. The solids were slurry washed three times, each with 20 mL water, for at least 15 minutes. The final cake was dried at 50 °C to 60 °C under reduced pressure to furnish 10 g of 5 containing less than 0.1% MeCN, 0.07% water, and 0.1% triflic acid (TfOH) by weight.
[0229] Alternatives to the above procedure employing MeCN and TfOH are as follows. Brominating agents: Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination Reagent Stoichiometry: Different amounts of the brominating reagent can be used, from about 0.8 equiv to about 2 equiv. Drying: A temperature range of about 10 °C to about 60 °C could be used for drying.
[0230] The impurity 5-a is was prepared and characterized as follows. 10 g of Compound 4 and sulfuric acid (35 mol%) were dissolved in MeOH (10 vol). The mixture was set to stir at 20 °C to 25 °C for 5-10 min and 2.0 equivalents of NBS were charged in one portion. The resulting yellow mixture was stirred for three days at 20-25 °C. The batch was concentrated under reduced pressure and the resulting solid was slurried in water at 95-100 °C for 3 hours. After a second overnight slurry in CH2CI2 at room temperature, the batch was filtered and dried to give a white solid 5-a (15.0 g, 78%). ¾ NMR (500 MHz, de-DMSO), 8.05 (2H, s, ArH), 3.40 (1H, br s, HO-Ar), 3.28 (3H, s, CH3); MS (ES‘) calc. 327/329/331; found
327/329/331; MP (DSC): 226 °C (onset 221 °C, 102 J/g); lit. 224-226 °C.
1.2: O-alkylation of 5 to produce 6
[0231] Compound 6 was prepared according to Scheme 7 below.
Scheme 7: O-alkylation of 5 to produce 6
[0232] Compound 5 (100 g, 398 mmol) and methyl ethyl ketone (MEK, 700 mL) were charged to the reaction vessel and agitated. Potassium carbonate (K2CO3, 325 mesh 82.56 g, 597 mmol) was then charged to the stirred reaction vessel at 15 °C to 25 °C.
Bromomethylcyclopropane (64.4 mL, 664 mmol) was charged to the reaction vessel over at least 1 hour, maintaining the temperature between 15 °C to 25 °C. MEK (200 mL) was added into the reactor and the reactor heated to 65 to 75 °C. The contents of the reaction vessel were stirred at 65 to 75°C for approximately 10 hours until reaction was complete by HPLC analysis. Water (3.0 vol, 3.0wt) was charged to the vessel maintaining the temperature at 65 to 75 °C. The batch was stirred at 65 to 75 °C. The phases were allowed to separate at 65°C to 75 °C and the lower aqueous phase was removed. Water (300 mL) was charged to the vessel maintaining the temperature at 65 °C to 75 °C. The batch was agitated for at least 10 minutes at 65 to 75 °C. The phases were allowed to separate at 65 °C to 75 °C and the lower aqueous phase was removed. The water wash was repeated once. The temperature was adjusted to 40 to 50°C. The mixture was concentrated to car. 500 mL under reduced pressure. The mixture was distilled under reduced pressure at up to 50 °C with MEK until the water content was <1.0% w/w. n-heptane (500mL) was charged to the vessel maintaining the temperature at 40 to 50 °C. The mixture was continuously distilled under vacuum with n-heptane (300mL), maintaining a 1L volume in the reaction vessel. Compound 6 seeds (0.0 lwt) were added at 40 to 50 °C. The mixture was continuously distilled under reduced pressure at up to 50 °C with n-heptane (300mL) while maintaining 1L volume in the reactor. The batch was cooled to 15 to 25 °C and aged for 2 hours. The product was collected by suction filtration. The filter cake was washed with a solution of 10% MEK in n-heptane (5vol) at 15 to 25°C. The filter cake was dried under reduced pressure at up to 40 °C under vacuum with nitrogen flow to afford 95g of 6. 1H NMR (500 MHz, de-DMSO) 8.07 (1H, d, 4J = 2.2 Hz, ArH), 7.86 (1H, d, J = 8.7 Hz, meta-ArH), 7.29 (1H, d, J = 8.8 Hz, ortho-AiK),
4.04 (2H, d, J = 6.9 Hz, OCH2CH), 3.21 (3H, s, CH3), 1.31-1.24 (1H, m, OCH), 0.62- 0.58 (2H, m, 2 x CHCHaHb), 0.40-0.37 (2H, m, 2 x CHC¾Hb); MS (ES+) calc. 305/307; found 305/307; MP: (DSC) 93 °C.
[0233] The following modifications of the above reaction, synthesis of 6 from 5, may be employed as well. Solvent: Different solvents could be used, for example acetone, methyl isobutyl ketone, ethyl acetate, isopropyl acetate, acetonitrile, or 2-methyl tetrahydrofuran. Reaction volume: Reaction volumes of 3 to 30 volumes with respect to 3 could be used. Base: Different inorganic bases, such as cesium carbonate or phosphate bases (sodium, potassium, or cesium) could be used. Also, organic bases, such as trimethylamine or diisopropyldiimide could be used. Base particle size: Different particle sizes of potassium carbonate from 325 mesh could be used. Reaction temperature: A lower temperature, such
as 50 °C could be used. A higher temperature, such as about 100 °C could be used. Any temperature above the boiling point of the solvent could be run in a pressure vessel.
Isolation: Different solvent ratios of MEK to n-heptane could be used. Different amounts of residual water can be left. Different amounts of seeds, from 0 to 50% could be used.
Seeding could take place later in the process and/or at a lower temperature. An un-seeded crystallization can be employed. A different isolation temperature, from 0 °C to 50 °C could be used. A different wash could be used, for example a different ratio of MEK to n-heptane. A different antisolvent from n-heptane could be used, such as hexane, pentane, or methyl tert-butyl ether. Alternatively, the batch could be solvent exchanged into a solvent where Compound 3 has a solubility of less than 100 mg/ml and isolated from this system. Drying: A temperature range of 10 to 60 °C could be used for drying.
[0234] Compound 10, shown below may also be formed as a result of O-alkylation of unreacted 4 present in product 5, or alternatively from or via a palladium mediated proteodesbromination or proteodesborylation in subsequent chemistry discussed in Example 1.3 below.
[0235] Preparation of methylsulfonylphenyl(cyclopropylmethyl) ether 10: Compound 4 (0.86 g, 5.0 mmol) and K2CO3 (1.04 g, 7.5 mmol) were slurried in acetone (17 mL, 20 vols). Cyclopropylmethyl bromide (0.73 mL, 7.5 mmol) was added in several small portions over ~1 minute and the reaction mixture heated to 50 °C for 48 hours, then cooled to 25 °C. Water (5.0 mL) was added with stirring and the acetone was evaporated on a rotary evaporator from which a fine white solid formed which was filtered off and returned to a vessel as a damp paste. A 1 : 1 mixture of MeOH/ water (8 mL) was added and heated to 40 °C with stirring. After 1 hour, the white solid was filtered off. Some residual solid was washed out with fresh water that was also rinsed through the cake, which was then isolated and left to air dry over the two days to give a dense white solid 10 (1.00 g, 88%). ¾ NMR (500 MHz, CDCb) 7.85
(2H, d, J = 8.8 Hz, RO-Ar ortho-H), 7.00 (2H, d, J = 8.8 Hz, RO-Ar meta- H), 3.87 (2H, d, J = 7.0 Hz, OCH2CH), 3.02 (3H, s, CHs), 1.34-1.23 (1H, m, OCH2CH), 0.72-0.60 (2H, m, 2 x CHCHflHb), 0.42-0.31 (2H, m, 2 x CHCH^.
1.3: Synthesis and Isolation Coupling Partner Boronic Ester 2
[0236] The final bond forming step to Compound 1 is a Suzuki-Miyaura coupling between Compounds 2 and 3, as shown in Scheme 3 below (Norio, M. and Suzuki, A., Chem. Rev., 95(7), 2457-2483 (1995)). Early studies demonstrated that the boronic ester of the isoquinolinone Compound 3-a had poor physical attributes and solid phase stability (Kaila, N. et al., J. Med Chem., 57: 1299-1322 (2014)). The pinacolatoboronate of the O-alkyl phenol, Compound 7, had acceptable solid phase stability and could be isolated via crystallization.
Scheme 3: Suzuki-Miyaura coupling between 2 and 3
[0237] Process robustness studies for the isolation of Compound 7, however, indicated that Compound 7 has poor solution stability, decomposing primarily to the proteodeborylated compound 10, as shown in Scheme 4 below. This was particularly problematic as the isolation process involved a solvent exchange from 2-MeTHF (2-methyl tetrahydrofuran) to iPrOAc (isopropyl acetate), which is not a fast unit operation on scale.
Scheme 4: Modification of 7
[0238] A search for a more stable boronic ester was undertaken. Early attempts targeted making N-methyliminodiacetic acid (MID A) boronate Compound 2-a (E. Gilis and M. Burke,“Multi step Synthesis of Complex Boronic Acids from Simple MIDA Boronates,” J Am. Chem. Soc., 750(43): 14084-14085 (2008)), however, all attempts resulted in product decomposition. Applicant then turned to a relatively obscure boronate formed by the addition of diethanolamine to Compound 7 (Bonin et al., Tetrahedron Lett., 52: 1132-1135 (2011)). Addition of diethanolamine to a solution of Compound 7 led to rapid ester formation and concomitant crystallization of Compound 2.
[0239] The discovery of boronic ester Compound 2 allowed for a simple, fast, high-yielding, high-purity process comprising the following procedure. Tetrahydrofuran (THF, 1500mL) was charged to a flask containing Compound 6 (100g, 328 mmol), bis(pinacolato)diboron (90.7g, 357 mmol) and cesium acetate (CsOAc, 158g, 822 mmol). The system was vacuum purged three times with nitrogen. Pd(PPh3)2Cl2 (13.8g, 20 mmol) was charged to the reaction and the system was vacuum purged three times with nitrogen. The reaction was then heated to 55 to 65°C.
[0240] The batch was stirred for approximately 8 hours until reaction was complete by HPLC analysis. The batch was cooled to 15 to 25 °C (target 20 °C ) and charged with silica gel (20g) and Ecosorb C-941 (20g). After lh, the mixture was filtered to remove solid. The residual solids were washed twice, each with THF (300mL). The filtrate and washes were combined. In a separate vessel, diethanolamine (34.5mL, 360 mmol) was dissolved in THF (250 mL). The diethanolamine solution in THF (25mL) was then charged to the batch. After 10 minutes, the batch was seeded with 2 (1 g) and aged for 1 to 2 hours. The remaining of the diethanolamine solution in THF was charged to the batch over at least 2 hours and the slurry was stirred for at least 2 hours. The product 2 was collected by suction filtration. The wet cake was washed thrice with THF (200mL). The material was dried under vacuum at 40 °C with nitrogen purge yielding 94.6g of 2.
[0241] The reaction to synthesize Compound 2 from Compound 6 described above may be modified as follows. Solvent: Different solvents from THF could be used, such as 1,4 dioxane or 2-methyltetrahydrofuran. Reaction volume: The reaction volume can be varied from 4 to 50 volumes with respect to compound 2. Catalyst and base: Different palladium catalyst and bases can be used for the borylation. Examples can be found in Chow et al., RSC Adv., 3 : 12518-12539 (2013). Borylation reaction temperature: Reaction temperatures from room temperature (20 °C) to solvent reflux can be used. Carbon/ Silica treatment:
The treatment can be performed without silica gel. The process can be performed without a carbon treatment. Different carbon sources from Ecosorb C-941 can be used. Different amounts of silica, from 0.01X to IX weight equivalents, can be used. Different amounts of Ecosorb C-941, from 0.01X to IX weight equivalents, can be used. Crystallization: A different addition rate of diethanolamine can be used. Different amounts of diethanolamine, from 1.0 to 3.0 molar equivalents can be used. A different cake wash with more or less THF can be used. Different amount of seeds from 0.0001X wt to 50X wt can be used.
Alternatively, the process can be unseeded. Drying: A temperature range of 10 °C to 60 °C could be used for drying.
[0242] The subsequent Suzuki-Miyaura coupling between Compounds 2 and 3 also proceeded well, providing over 20 kg of crude compound 1 with an average molar yield of 80% and LCAP of 99.7%.
1.4: Synthesis of Coupling Partner 3
[0243] Cross-coupling partner 3 was prepared by two different processes corresponding to Schemes 8 and 9 shown below.
Scheme 8: Process A for preparation of 3
[0244] According to Process A, Compound 9 (100g, 628 mmol) was dissolved in acetonitrile (450 mL) at room temperature. In a separate vessel, N-bromosuccinimide (NBS, 112g, 628 mmol) was suspended in acetonitrile (1 L). Compound 9 in acetonitrile was charged to the NBS slurry over at least 45 minutes. The contents of the reaction vessel were warmed to 45 °C to 55 °C and the batch stirred until the reaction was complete by HPLC analysis. The batch was cooled to 35 °C to 45 °C and ensured dissolution. Norit SX plus carbon (lOg) was charged to the mixture and the reaction mixture adjusted to 55 °C to 60 °C. The mixture was stirred at 55 °C to 60 °C for about lh and the mixture filtered at 55 °C to 60 °C to remove solids. The solids were washed with acetonitrile (500mL) at 55 °C to 60 °C. The volume of the combined filtrate was reduced to 900 mL by distilling off acetonitrile under reduced pressure. The batch with Compound 3 (lg) and stirred at 35 °C to 45 °C for at least 60 minutes. The contents of the reaction vessel were cooled to 15 °C to 25 °C over at least 1 hour. Water (2000 mL) was charged to the reaction vessel over at least 90 minutes and the slurry aged for at least 60 minutes. The product was collected by suction filtration. The cake was washed with a premixed 5% solution of acetonitrile in water (300mL). The wet cake was dried under vacuum at 40 °C with nitrogen purge. Yield: 120g of 3.
[0245] The above procedure, Process A for this synthesis of 3, may be practiced with alternative reagents and conditions as follows. Solvents: Alternative solvents could be used. Examples include chlorinated solvents, such as methylene chloride, chloroform or 1,2 dichloroethane, and non-chlorinated solvents such as tetrahydrofuran, or 2-methyltetrahydrofuran. Reaction concentration: The reaction concentration can be varied from 2X vol to 40 X vol (with respect to Compound 9). Brominating agents: Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination reagent Stoichiometry: Different amounts of the brominating reagent can be used, from 0.8 equiv to 2 equiv. Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used. The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 °C to 60 °C could be used for drying.
Scheme 9: Process B for preparation of 3
[0246] According to Process B, Compound 3 can be formed starting from 8 via non-isolated compound 9 as follows. Compound 8 (80 g, 55 mmol), cesium carbonate (CS2CO3, 215 g, 66 mmol), and acetonitrile (800 mL) were charged to the reactor. The temperature was adjusted from 15 to 25 °C and iodomethane charged to the reactor (Mel, 86 g, 0.61 mol) while maintaining a batch temperature below 25 °C. The batch was heated to 40 °C and agitated for 10 hours to form Compound 9. The batch was cooled to 25 °C, filtered into a fresh reactor to remove solids, and the solids washed twice with acetonitrile. The combined organic layers were concentrated via atmospheric distillation to about 320 mL.
[0247] In a separate reactor N-bromosuccinimide (NBS, 98.1 g, 0.55 mol) was charged to acetonitrile (800 mL) and agitated. The batch containing Compound 9 was transferred to the NBS solution while maintaining a batch temperature of 15 to 25 °C. The batch was heated to 45 to 55 °C and agitated for at least 4 hours to allow for reaction completion to Compound 3. Upon reaction completion, Norit SX Plus activated carbon (8 g) was charged, and agitated at 45 to 55 °C for one hour. The batch was filtered into a fresh vessel, the Norit SX plus cake was washed with 400 ml of 45 to 55 °C acetonitrile. The acetonitrile layers were combined, cooled to 35 to 45 °C, and distilled under reduced pressure to 720 mL. The batch was adjusted to a temperature of 40 °C, charged with Compound 3 seeds (0.8 g), agitated for one hour, cooled to 15 to 25 °C over at least on hour, then charged with water (1600 mL) over at least two hours. The mixture was agitated for an additional one to two hours, filtered, the cake washed with a premixed 5% solution of acetonitrile in water (240 mL). The wet cake was dried under vacuum at 40°C with nitrogen purge. Yield: 52 g of 3.
[0248] Process B to synthesize Compound 3, described above, may be modified as follows. Solvents: Alternative solvents could be used. Examples include chlorinated solvents, such as methylene chloride, chloroform or 1,2 dichloroethane, and non-chlorinated solvents such as tetrahydrofuran, or 2-methyltetrahydrofuran. Reaction concentration: The reaction concentration can be varied from 2X vol to 40 X vol (with respect to Compound 8).
Alkylating reagent: Alternative methylating reagents to methyl iodide can be used such as dimethylsulfate. Alkylating reagent stoichiometry: 1 to 10 molar equivalents of methyl iodide may be used. Base: Different inorganic bases, such as potassium carbonate or phosphate bases (sodium, potassium, or cesium) could be used. Brominating agents:
Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination reagent stoichiometry: Different amounts of the brominating reagent can be used, from 0.8 equiv to 2 equiv. Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used. Seeding levels from 0.0001% to 50% can be used. The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying.
1.5: Cross-coupling of 2 and 3 to Produce Target Compound 1
[0249] 1 is synthesized by Suzuki cross-coupling of 3 and 2 according to Scheme 10 and as described below.
Scheme 10: Synthesis of 1
[0250] Acetonitrile (1.6L) was charged to a mixture of Compound 2 (156.7g, 460 mmol), Compovmd 3 (lOOg, 420 mmol) and potassium phosphate tribasic (223 g, l.OSmol). Agitation was begun and water (400mL) charged to the batch. The system was vacuum purged three times with nitrogen and charged with Pd(PPh3)2Cl2 (2.9g, 4 mmol) and the system vacuum
purged three times with nitrogen. The batch was heated to 65 to 75°C and contents stirred for at least 16 hours until reaction was complete by HPLC analysis. The batch was cooled to 60 to 70°C, agitation halted and the mixture allowed to settle. The bottom aqueous layer was removed. Water (150mL) and acetonitrile (700mL) were charged at 60 to 70°C. Ecosorb C-941 (15g) and Celite (lOg) were charged to the reaction vessel at 60 to 70°C. After lh, the mixture was filtered to remove solids. The solids were washed twice each with 18% water in acetonitrile (500 mL) at 60 to 70°C. The filtrates were combined and concentrated under atmospheric pressure to a final volume of 1.5L. The batch was cooled to 60 to 65°C and seeded with Compound 1 (1 g). After lh, water (500 mL) was charged over at least 1 hour at 60 to 65°C. The slurry was cooled to 15 to 25°C over 4 hours. The product was collected by suction filtration. The wet cake was washed with 45% water in acetonitrile (500mL) twice. The product was dried under vacuum at 40°C with nitrogen purge. Yield: 139g of 1.
[0251] The above procedure for coupling Compound 3 and Compound 2 to produce
Compound 1 may be modified in any of the ways that follow. Reaction solvents: Different reaction solvents from acetonitrile can be used, including tetrahydrofuran, 2-methyl tetrahydrofuran, toluene, and isopropanol. Boronic ester: Different boronic esters from Compound 2 can be used, including pinacolato ester compound 7, and the free boronic acid of Compound 2. Examples of boronic esters can be found in Lennox, Alister, J.J., Lloyd-Jones, Guy C. Chem. Soc. Rev., 2014, 43, 412. Carbon treatment: Different carbon treatments from Ecosorb C-941 could be used. Different amounts of carbon, from 0.01 to 0.5X weight can be used. The carbon can be eliminated. Different amounts of Celite, from 0.01 to 0.5X weight can be used. Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used. The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying. Catalysts: Different metal and ligand combination could be used. Examples of metal/ligand combinations can be found in Maluenda, Irene; Navarro, Oscar, Molecules, 2015, 20, 7528. Various catalysts can be including: XPhos-3G (cas# 1445085-55-1);
catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
1.6: Crystallization of 1
[0252] The final isolation of Compound 1 requires a polish filtration. For this, the batch must be completely soluble. Unfortunately, Compound 1 has low solubility in almost all
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Class 3 and common Class 2 (e.g. THF, MeCN) solvents (ICH
Harmonized Guideline“Impurities: Guideline for Residual Solvents Q3C(R6)” October 20, 2016). A reasonable solubility was obtained in a warm MeCN-water mix, but this is not an optimal system (requires a heated filtration, MeCN has a residual solvent limit of only 410 ppm). Additional solvents with reasonable solubility (>50 mg/ml) include N-methyl-2- pyrrolidone (NMP) and dimethylacetamide (DMAc); but the development of isolations from these solvents required large volumes and raised residual solvent limit concerns (530 ppm or less for NMT and 1090 ppm or less for DMAc).
catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
1.6: Crystallization of 1
[0252] The final isolation of Compoxmd 1 requires a polish filtration. For this, the batch must be completely soluble. Unfortunately, Compound 1 has low solubility in almost all
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Class 3 and common Class 2 (e.g. THF, MeCN) solvents (ICH
Harmonized Guideline“Impurities: Guideline for Residual Solvents Q3C(R6)” October 20, 2016). A reasonable solubility was obtained in a warm MeCN-water mix, but this is not an optimal system (requires a heated filtration, MeCN has a residual solvent limit of only 410 ppm). Additional solvents with reasonable solubility (>50 mg/ml) include N-methyl-2- pyrrolidone (NMP) and dimethylacetamide (DMAc); but the development of isolations from these solvents required large volumes and raised residual solvent limit concerns (530 ppm or less for NMT and 1090 ppm or less for DMAc).
[0253] Formic acid is one ICH Class 3 solvent in which Compound 1 is highly soluble, having a solubility greater than 250 mg/ml at 20 °C. The solubility curve of Compound 1 in formic acid-Water is quite steep (see Figure 7), which enables a volumetrically efficient process.
[0254] Initial attempts to recrystallize crude Compound 1 involved dissolving in formic acid, polish filtering, and charging polish filtered water to about 20% supersaturation, followed by seeding with the thermodynamically most stable form (Form 1), followed by slow addition of water to the final solvent ratio, filtration, washing, and drying. Applicant observed that during the initial water charge, if the batch self-seeded it formed a thick slurry. X-ray diffraction (XRD), differential scanning calorimetry (DSC), and photomicroscopy demonstrated that a metastable form was produced. Once seeded with Form 1, the batch converted to the desired form (Form 1) prior to the addition of the remaining water. This process worked well during multiple lab runs, consistently delivering the desired form and purity with about 85% yield.
[0255] Unfortunately, upon scale-up, the batch did not convert to Form 1 after seeding. Additional water was charged and the batch began to convert to the desired form (mix of Form 1 and the metastable form by X-ray powder diffraction (XRPD)). When additional water was charged, the XRPD indicated only the metastable form. After a few hours with no change, Applicant continued the water charge to the final solvent ratio, during which time the batch eventually converted to Form 1. This process is summarized in Figure 8.
[0256] It was subsequently found by closer analysis of the plant and laboratory retains that a new metastable form was formed during scale up, with a similar, but different XRPD pattern. This form (metastable B) could be reproduced in the laboratory, but only when the batch has a high formic acid:water ratio and is seeded with Form 1. Without Form 1 seeds, metastable A is the kinetic form. Both metastable forms converted to Form 1 with additional water and/or upon drying, leading Applicant to believe that the metastable forms are formic acid solvates. These findings are summarized in Figure 9.
[0257] While there is little risk in not being able to control the final form, there is a risk of forming a difficult-to-stir slurry which can lead to processing issues. The crystallization procedure was therefore modified to keep a constant formic acid-water ratio. This was performed by charging 2.4X wt. formic acid and 1.75X wt. water (final solvent composition)
to the crystallizer with 0.03X wt. Form 1 seeds, and performing a simultaneous addition of Compound 1 in 6. IX wt. formic acid and 4.4X wt. water. The batch filtered easily and was washed with formic acid/water, then water, and dried under reduced pressure to yield 8.9 kg of Compound 1 (92% yield) with 99.85% LCAP and N.D. formic acid.
Example 2: Exemplary high throughput experimentation reaction
[0258] The following procedure is an exemplary high throughput experimentation reaction.
[0259] An overview of the reaction is shown below in Scheme 5:
Scheme 5: Reaction conditions tested for cross-coupling reaction of 2 and 3
[0260] Pd catalysts were dosed into the 24-well reactor vial as solutions (100 pL of 0.01 M solution in tetrahydrofuran (THF) or dichloroethane (DCE) depending upon the solubility of the ligand). Plates of these ligands are typically dosed in advance of the reaction, the solvent is removed by evacuation in an evaporative centrifuge and plates are stored in the glovebox. The catalysts screened in the coupling are the following: XPhos, SPhos, CataCXium A, APhos, P(Cy)3, PEPPSI-IPent. For the first five ligands, these were initially screened as the Buchwald Pd G2/G3 precatalysts.
[0261] To the plates was then added a stock solution of Compound 3 (10 pmol) and Compound 2 (12 pmol) dissolved in the following solvents: dimethylformamide (DMF),
tetrahydrofuran (THF), butanol (/r-BuOH), and toluene. The base was then added as a stock solution (30 mmol) in 20 mL of water.
[0262] A heatmap summarizing catalyst performance is shown in Figs. 10A and 10B. High performance liquid chromatography (HPLC) yields for this screening span from <5% up to -85%. Larger circles indicate higher yield. Lighter circles indicate higher cleanliness.
[0263] A similarly designed screening of base and solvent also indicate that a range of alcoholic solvents (methanol, ethanol, propanol, 2-butanol, 2-propanol, and /-amyl alcohol) are also all viable in this coupling chemistry. Bases such as potassium phosphate, potassium carbonate, potassium acetate, and potassium hydroxide were all successful in achieving the coupling. Fig. 10B shows a heatmap with HPLC yields ranging from -50 – 95%. Larger, darker circles indicate higher yield.
[0264] This chemistry from microvial screening has been scaled to a laboratory process. To a 3 -necked jacketed 250 mL flask equipped with overhead stirring, nitrogen inlet, and thermocouple was added Compound 3 (1.0 eq, 4.00 grams), Compound 2 (1.2 eq, 1.71 x wt), potassium carbonate (3.0 eq, 1.74 x wt). The reactor was inerted three times and then degassed 2-propanol (24 x vol.) followed by degassed water (6 x vol) was then added.
Stirring was then initiated at 300 rpms. The reactor was then stirred and blanketed with nitrogen for 1 hour. The catalyst was then added (0.01 eq, 0.028 x wt) and stirring continued (300 rpms) and the reactor was heated into the Tj = 65 °C.
[0265] After 2 hours, with full conversion confirmed analytically, trioctylphosphine (0.1 eq, 0.16 x wt) dosed, and reaction mixture allowed to cool slowly to room temperature hours.
The reaction mixture was then filtered, washed with 2-propanol (4 x vol), 2-propanol: water (4: 1, 4 x vol), and then with water (4 x vol). Note: If 2 is dimer present in cake, an additional ethyl acetate (EtOAc) wash (4 x vol) can be added for purging. The cake was then transferred to a vacuum oven to dry overnight at 40 °C, -40 cm Hg, under nitrogen flow. After transfer to a bottle, 6.03 grams of 1 were isolated, 98.6% assay, 91% overall yield.
Scheme 6: Alternative reagents and solvents for cross-coupling
[0266] Based on the previously delineated results, it was expected that a variety of monodentate (PPI13 [triphenylphosphine], PBu3 [tributylphosphine], etc) and bidentate phosphines (dppf [1,1 ‘-bis(diphenylphosphino)ferrocene], BINAP [2,2 -bis(diphenylphosphino)- 1 , 1 -binaphthyl], Xantphos [4,5-bis(diphenylphosphino)-9,9-dimethylxanthene], dppe [l,2-bis(diphenylphosphino)ethane], etc) ligated to any number of Pd sources (Pd halides, Pd(H) precatalyts, Pd(0) sources) could reasonably be employed to arrive at the Compound 1 crude material. A range of organic solvents ranging from non-polar (heptane, benzene), protic (alcohols), polar aprotic (dimethylsulfoxide, dimethylformamide, dimethylacetamide, acetonitrile) as well as a variety of esters and ketones (acetone, 2-butanone, ethylacetate) should also serve as effective solvents for this reactivity. Finally, inorganic bases of varying strength (phosphates, carbonates, acetates, etc) along with organic variants such as triethylamine, l,8-diazabicyclo(5.4.0)undec-7-ene, and others in a wide pKa range are viable as stoichiometric basic additives.
Example 3: Exemplary Compound 5 process
[0267] The purpose of this example was to describe an exemplary process for making Compound 5.
[0268] Charge 4 (lOg, 58mmol) and acetonitrile (lOOmL) to a reaction vessel and start the stirrer. Adjust the batch to -18 °C to -22 °C (target -20 °C). Charge triflic acid (5.5mL, 62mmol) to the batch maintaining -10 °C to -25 °C (target -20 °C). Stir the batch at -10 °C to -25 °C (target -20 °C) for 10 to 20 minutes. Charge NBS (11.38g, 64mmol) to the batch at -10 °C to -25 °C (target -20 °C) and stir for ca. 30 min at -10 °C to -25 °C (target -20 °C). Warm the batch to 20 °C over 3-4 hours (reaction will occur when internal temp is between 5 °C and 15 °C). Stir the batch at 15 °C to 25 °C (target 20 °C) for approximately 1 hour and sample for reaction completion.
[0269] If Compound 4 relative to Compound 5 is more than 5%:
[0270] Cool the bath to -5 °C to -15 °C (target -10 °C) (cooling below 0 °C to ensure selectivity). Charge NBS to the batch according to the follow formula: Mass of NBS = (% Compound 4 x lOg). Warm the batch to 20 °C over 1-2 hours. Stir the batch at 15 °C to 25 °C (target 20 °C) for approximately 1 hour and check reaction for completion. Proceed to next line.
[0271] If Compound 4 relative to Compound 5 is less than 5%:
[0272] Warm the batch to 40 °C to 50 °C (target 48 °C). Concentrate the batch under reduced pressure to a final volume of ~40mL. Cool the batch to -15 °C to -5 °C (target -10 °C) and stir for ca. lh. Filter the batch by suction filtration. Slurry wash the filter cake with purified water (3 x 20mL) at 15 °C to 25 °C (target 20 °C) for 10 to 15 minutes each wash. Remove a sample of the filter cake for analysis by ¾ NMR. Continue washing cake until the residual succimide is below 1.0%mol% relative to 5. Dry the filter cake at up to 60°C under vacuum and nitrogen purge. Analyse the 5 by HPLC analysis (97%w/w to 99%w/w). Expected yield: 60-85% theory (90-110% w/w).
Example 4: Purification of Compound 1 (CC-90010) by crystallization from formic acid and water.
[0273] This example describes a method for the purification of Compound 1 by
crystallization from formic acid and water. Also detailed are methods for obtaining three different polymorphs of Compound 1, including the most stable form, Form 1.
[0274] Figure 11 shows XH NMR of Compound 1 (CC-90010). Solvent: d6DMSO; and Figure 12 shows microscopy of Compound 1 (CC-90010) Form I. Figure 13 shows XRPD of Compound 1 (CC-90010) Form I, with peak information detailed in Table 6:
Sacituzumab Govitecan is an antibody drug conjugate containing the humanized monoclonal antibody, hRS7, against tumor-associated calcium signal transducer 2 (TACSTD2 or TROP2) and linked to the active metabolite of irinotecan, 7-ethyl-10-hydroxycamptothecin (SN-38), with potential antineoplastic activity. The antibody moiety of sacituzumab govitecan selectively binds to TROP2. After internalization and proteolytic cleavage, SN-38 selectively stabilizes topoisomerase I-DNA covalent complexes, resulting in DNA breaks that inhibit DNA replication and trigger apoptosis. TROP2, also known as epithelial glycoprotein-1 (EGP-1), is a transmembrane calcium signal transducer that is overexpressed by a variety of human epithelial carcinomas; this antigen is involved in the regulation of cell-cell adhesion and its expression is associated with increased cancer growth, aggressiveness and metastasis.
FDA Approves Trodelvy®, the First Treatment for Metastatic Triple-Negative Breast Cancer Shown to Improve Progression-Free Survival and Overall Survival
– Trodelvy Significantly Reduced the Risk of Death by 49% Compared with Single-Agent Chemotherapy in the Phase 3 ASCENT Study –
– Trodelvy is Under Regulatory Review in the EU and in the United Kingdom, Canada, Switzerland and Australia as Part of Project Orbis–April 07, 2021 07:53 PM Eastern Daylight Time
FOSTER CITY, Calif.–(BUSINESS WIRE)–Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the U.S. Food and Drug Administration (FDA) has granted full approval to Trodelvy® (sacituzumab govitecan-hziy) for adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease. The approval is supported by data from the Phase 3 ASCENT study, in which Trodelvy demonstrated a statistically significant and clinically meaningful 57% reduction in the risk of disease worsening or death (progression-free survival (PFS)), extending median PFS to 4.8 months from 1.7 months with chemotherapy (HR: 0.43; 95% CI: 0.35-0.54; p<0.0001). Trodelvy also extended median overall survival (OS) to 11.8 months vs. 6.9 months (HR: 0.51; 95% CI: 0.41-0.62; p<0.0001), representing a 49% reduction in the risk of death.
Trodelvy is directed to the Trop-2 receptor, a protein frequently expressed in multiple types of epithelial tumors, including TNBC, where high expression is associated with poor survival and relapse. Prior to the FDA approval of Trodelvy, patients with previously treated metastatic TNBC had few treatment options in this high unmet-need setting. The FDA granted accelerated approval to Trodelvy in April 2020 based on objective response rate and duration of response results in a Phase 1/2 study. Today’s approval expands the previous Trodelvy indication to include treatment in adult patients with unresectable locally advanced or metastatic TNBC who have received two or more prior systemic therapies, at least one of them for metastatic disease.
“Women with triple-negative breast cancer have historically had very few effective treatment options and faced a poor prognosis,” said Aditya Bardia, MD, MPH, Director of Breast Cancer Research Program, Mass General Cancer Center and Assistant Professor of Medicine at Harvard Medical School, and global principal investigator of the ASCENT study. “Today’s FDA approval reflects the statistically significant survival benefit seen in the landmark ASCENT study and positions sacituzumab govitecan-hziy as a potential standard of care for pre-treated TNBC.”
“A metastatic TNBC diagnosis is frightening. As an aggressive and difficult-to-treat disease, it’s a significant advance to have an FDA-approved treatment option with a proven survival benefit for patients with metastatic disease that continues to progress,” said Ricki Fairley, Founder and CEO of Touch, the Black Breast Cancer Alliance. “For far too long, people with metastatic TNBC had very few treatment options. Today’s news continues the progress of bringing more options to treat this devastating disease.”
Among all patients evaluable for safety in the ASCENT study (n=482), Trodelvy had a safety profile consistent with the previously approved FDA label. The most frequent Grade ≥3 adverse reactions for Trodelvy compared to single-agent chemotherapy were neutropenia (52% vs. 34%), diarrhea (11% vs. 1%), leukopenia (11% vs. 6%) and anemia (9% vs. 6%). Adverse reactions leading to treatment discontinuation occurred in 5% of patients receiving Trodelvy.
“Today’s approval is the culmination of a multi-year development program and validates the clinical benefit of this important treatment in metastatic TNBC,” said Merdad Parsey, MD, PhD, Chief Medical Officer, Gilead Sciences. “Building upon this milestone, we are committed to advancing Trodelvy with worldwide regulatory authorities so that, pending their decision, Trodelvy may become available to many more people around the world who are facing this difficult-to-treat cancer.”
Regulatory submissions for Trodelvy in metastatic TNBC have been filed in the United Kingdom, Canada, Switzerland and Australia as part of Project Orbis, an initiative of the FDA Oncology Center of Excellence (OCE) that provides a framework for concurrent submission and review of oncology products among international partners, as well as in Singapore through our partner Everest Medicines.The European Medicines Agency has also validated a Marketing Authorization Application for Trodelvy in the European Union. All filings are based on data from the Phase 3 ASCENT study.
Trodelvy Boxed Warning
The Trodelvy U.S. Prescribing Information has a BOXED WARNING for severe or life-threatening neutropenia and severe diarrhea; see below for Important Safety Information.
About Trodelvy
Trodelvy (sacituzumab govitecan-hziy) is a first-in-class antibody and topoisomerase inhibitor conjugate directed to the Trop-2 receptor, a protein frequently expressed in multiple types of epithelial tumors, including metastatic triple-negative breast cancer (TNBC), where high expression is associated with poor survival and relapse.
Trodelvy is also being developed as an investigational treatment for metastatic urothelial cancer, hormone receptor-positive/human epidermal growth factor receptor 2-negative (HR+/HER 2-) metastatic breast cancer and metastatic non-small cell lung cancer. Additional evaluation across multiple solid tumors is also underway.
About Triple-Negative Breast Cancer (TNBC)
TNBC is an aggressive type of breast cancer, accounting for approximately 15% of all breast cancers. The disease is diagnosed more frequently in younger and premenopausal women and is more prevalent in African American and Hispanic women. TNBC cells do not have estrogen and progesterone receptors and have limited HER 2. Medicines targeting these receptors therefore are not typically effective in treating TNBC.
About the ASCENT Study
The Phase 3 ASCENT study, an open-label, active-controlled, randomized confirmatory trial, enrolled more than 500 patients with relapsed/refractory metastatic triple-negative breast cancer (TNBC) who had received two or more prior systemic therapies (including a taxane), at least one of them for metastatic disease. Patients were randomized to receive either Trodelvy or a chemotherapy chosen by the patients’ treating physicians. The primary efficacy outcome was progression-free survival (PFS) in patients without brain metastases at baseline, as measured by a blinded, independent, centralized review using RECIST v1.1 criteria. Additional efficacy measures included PFS for the full population (all patients with and without brain metastases) and overall survival (OS). More information about ASCENT is available at http://clinicaltrials.gov/show/NCT02574455.
Important Safety Information for Trodelvy
BOXED WARNING: NEUTROPENIA AND DIARRHEA
Severe, life-threatening, or fatal neutropenia may occur. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Consider G-CSF for secondary prophylaxis. Initiate anti-infective treatment in patient with febrile neutropenia without delay.
Severe diarrhea may occur. Monitor patients with diarrhea and give fluid and electrolytes as needed. Administer atropine, if not contraindicated, for early diarrhea of any severity. At the onset of late diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤ Grade 1 and reduce subsequent doses.
CONTRAINDICATIONS
Severe hypersensitivity to TRODELVY
WARNINGS AND PRECAUTIONS
Neutropenia: Dose modifications may be required due to neutropenia. Neutropenia occurred in 62% of patients treated with TRODELVY, leading to permanent discontinuation in 0.5% of patients. Grade 3-4 neutropenia occurred in 47% of patients. Febrile neutropenia occurred in 6%.
Diarrhea: Diarrhea occurred in 64% of all patients treated with TRODELVY. Grade 3 diarrhea occurred in 12% of patients. Neutropenic colitis occurred in 0.5% of patients. Withhold TRODELVY for Grade 3-4 diarrhea and resume when resolved to ≤ Grade 1. At onset, evaluate for infectious causes and if negative, promptly initiate loperamide, 4 mg initially followed by 2 mg with every episode of diarrhea for a maximum of 16 mg daily. Discontinue loperamide 12 hours after diarrhea resolves. Additional supportive measures (e.g., fluid and electrolyte substitution) may also be employed as clinically indicated. Patients who exhibit an excessive cholinergic response to treatment can receive appropriate premedication (e.g., atropine) for subsequent treatments.
Hypersensitivity and Infusion-Related Reactions: TRODELVY can cause severe and life-threatening hypersensitivity and infusion-related reactions, including anaphylactic reactions. Hypersensitivity reactions within 24 hours of dosing occurred in 37% of patients. Grade 3-4 hypersensitivity occurred in 1% of patients. The incidence of hypersensitivity reactions leading to permanent discontinuation of TRODELVY was 0.4%. Pre-infusion medication is recommended. Observe patients closely for hypersensitivity and infusion-related reactions during each infusion and for at least 30 minutes after completion of each infusion. Medication to treat such reactions, as well as emergency equipment, should be available for immediate use.
Nausea and Vomiting: Nausea occurred in 67% of all patients treated with TRODELVY. Grade 3-4 nausea occurred in 5% of patients. Vomiting occurred in 40% of patients and Grade 3-4 vomiting occurred in 3% of these patients. Premedicate with a two or three drug combination regimen (e.g., dexamethasone with either a 5-HT3 receptor antagonist or an NK-1 receptor antagonist as well as other drugs as indicated) for prevention of chemotherapy-induced nausea and vomiting (CINV). Withhold TRODELVY doses for Grade 3 nausea or Grade 3-4 vomiting and resume with additional supportive measures when resolved to Grade ≤ 1. Additional antiemetics and other supportive measures may also be employed as clinically indicated. All patients should be given take-home medications with clear instructions for prevention and treatment of nausea and vomiting.
Increased Risk of Adverse Reactions in Patients with Reduced UGT1A1 Activity: Individuals who are homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia and may be at increased risk for other adverse reactions with TRODELVY. The incidence of Grade 3-4 neutropenia in genotyped patients was 69% in patients homozygous for the UGT1A1*28, 48% in patients heterozygous for the UGT1A1*28 allele and 46% in patients homozygous for the wild-type allele. The incidence of Grade 3-4 anemia in genotyped patients was 24% in patients homozygous for the UGT1A1*28 allele, 8% in patients heterozygous for the UGT1A1*28 allele, and 10% in patients homozygous for the wild-type allele. Closely monitor patients with known reduced UGT1A1 activity for adverse reactions. Withhold or permanently discontinue TRODELVY based on severity of the observed adverse reactions in patients with evidence of acute early-onset or unusually severe adverse reactions, which may indicate reduced UGT1A1 function.
Embryo-Fetal Toxicity: Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. TRODELVY contains a genotoxic component, SN-38, and targets rapidly dividing cells. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose.
ADVERSE REACTIONS
In the ASCENT study (IMMU-132-05), the most common adverse reactions (incidence ≥25%) were nausea, neutropenia, diarrhea, fatigue, alopecia, anemia, vomiting, constipation, rash, decreased appetite, and abdominal pain. The most frequent serious adverse reactions (SAR) (>1%) were neutropenia (7%), diarrhea (4%), and pneumonia (3%). SAR were reported in 27% of patients, and 5% discontinued therapy due to adverse reactions. The most common Grade 3-4 lab abnormalities (incidence ≥25%) in the ASCENT study were reduced hemoglobin, lymphocytes, leukocytes, and neutrophils.
DRUG INTERACTIONS
UGT1A1 Inhibitors: Concomitant administration of TRODELVY with inhibitors of UGT1A1 may increase the incidence of adverse reactions due to potential increase in systemic exposure to SN-38. Avoid administering UGT1A1 inhibitors with TRODELVY.
UGT1A1Inducers: Exposure to SN-38 may be substantially reduced in patients concomitantly receiving UGT1A1 enzyme inducers. Avoid administering UGT1A1 inducers with TRODELVY
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer. Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
The most common side effects are nausea, neutropenia, diarrhea, fatigue, anemia, vomiting, alopecia (hair loss), constipation, decreased appetite, rash and abdominal pain.[1][2] Sacituzumab govitecan has a boxed warning about the risk of severe neutropenia (abnormally low levels of white blood cells) and severe diarrhea.[1][2] Sacituzumab govitecan may cause harm to a developing fetus or newborn baby.[1] Women are advised not to breastfeed while on sacituzumab govitecan and 1 month after the last dose is administered.[3]
Sacituzumab govitecan is a conjugate of the humanized anti-Trop-2monoclonal antibody linked with SN-38, the active metabolite of irinotecan.[5] Each antibody having on average 7.6 molecules of SN-38 attached.[6] SN-38 is too toxic to administer directly to patients, but linkage to an antibody allows the drug to specifically target cells containing Trop-2.
Sacituzumab govitecan is a Trop-2-directed antibody and topoisomerase inhibitor drug conjugate, meaning that the drug targets the Trop-2 receptor that helps the cancer grow, divide and spread, and is linked to topoisomerase inhibitor, which is a chemical compound that is toxic to cancer cells.[1] Approximately two of every ten breast cancer diagnoses worldwide are triple-negative.[1] Triple-negative breast cancer is a type of breast cancer that tests negative for estrogen receptors, progesterone receptors and human epidermal growth factor receptor 2 (HER2) protein.[1] Therefore, triple-negative breast cancer does not respond to hormonal therapy medicines or medicines that target HER2.[1]
Development
Immunomedics announced in 2013, that it had received fast track designation from the US Food and Drug Administration (FDA) for the compound as a potential treatment for non-small cell lung cancer, small cell lung cancer, and metastatic triple-negative breast cancer. Orphan drug status was granted for small cell lung cancer and pancreatic cancer.[7][8] In February 2016, Immunomedics announced that sacituzumab govitecan had received an FDA breakthrough therapy designation (a classification designed to expedite the development and review of drugs that are intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition) for the treatment of patients with triple-negative breast cancer who have failed at least two other prior therapies for metastatic disease.[9][10]
History
Sacituzumab govitecan was added to the proposed INN list in 2015,[11] and to the recommended list in 2016.[12]
Sacituzumab govitecan-hziy was approved for use in the United States in April 2020.[1][13][14][2]
Sacituzumab govitecan-hziy was approved based on the results of IMMU-132-01, a multicenter, single-arm clinical trial (NCT01631552) of 108 subjects with metastatic triple-negative breast cancer who had received at least two prior treatments for metastatic disease.[1][14][2] Of the 108 patients involved within the study, 107 were female and 1 was male.[15] Subjects received sacituzumab govitecan-hziy at a dose of 10 milligrams per kilogram of body weight intravenously on days one and eight every 21 days.[14][15] Treatment with sacituzumab govitecan-hziy was continued until disease progression or unacceptable toxicity.[15] Tumor imaging was obtained every eight weeks.[14][2] The efficacy of sacituzumab govitecan-hziy was based on the overall response rate (ORR) – which reflects the percentage of subjects that had a certain amount of tumor shrinkage.[1][14] The ORR was 33.3% (95% confidence interval [CI], 24.6 to 43.1). [1][14][15] Additionally, with the 33.3% of study participants who achieved a response, 2.8% of patients experienced complete responses.[15] The median time to response in patients was 2.0 months (range, 1.6 to 13.5), the median duration of response was 7.7 months (95% confidence interval [CI], 4.9 to 10.8), the median progression free survival was 5.5 months, and the median overall survival was 13.0 months.[15] Of the subjects that achieved an objective response to sacituzumab govitecan-hziy, 55.6% maintained their response for six or more months and 16.7% maintained their response for twelve or more months.[1][14]
^ World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): proposed INN: list 113”. WHO Drug Information. 29 (2): 260–1. hdl:10665/331080.
^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 75”. WHO Drug Information. 30 (1): 151–3. hdl:10665/331046.
Dostarlimab, sold under the brand name Jemperli, is a monoclonal antibody medication used for the treatment of endometrial cancer.[1][2][3][4]
The most common adverse reactions (≥20%) were fatigue/asthenia, nausea, diarrhea, anemia, and constipation.[1][2] The most common grade 3 or 4 adverse reactions (≥2%) were anemia and transaminases increased.[1][2]
Dostarlimab is a programmed death receptor-1 (PD-1)–blocking antibody.[1][2]
Dostarlimab was approved for medical use in the United States in April 2021.[1][2][5]
NAME
DOSAGE
STRENGTH
ROUTE
LABELLER
MARKETING START
MARKETING END
Jemperli
Injection
50 mg/1mL
Intravenous
GlaxoSmithKline LLC
2021-04-22
Not applicable
Medical uses
Dostarlimab is indicated for the treatment of adults with mismatch repair deficient (dMMR) recurrent or advanced endometrial cancer, as determined by an FDA-approved test, that has progressed on or following prior treatment with a platinum-containing regimen.[1][2]
On April 22, 2021, the Food and Drug Administration granted accelerated approval to dostarlimab-gxly (Jemperli, GlaxoSmithKline LLC) for adult patients with mismatch repair deficient (dMMR) recurrent or advanced endometrial cancer, as determined by an FDA-approved test, that has progressed on or following a prior platinum-containing regimen.
Efficacy was evaluated based on cohort (A1) in GARNET Trial (NCT02715284), a multicenter, multicohort, open-label trial in patients with advanced solid tumors. The efficacy population consisted of 71 patients with dMMR recurrent or advanced endometrial cancer who progressed on or after a platinum-containing regimen. Patients received dostarlimab-gxly, 500 mg intravenously, every 3 weeks for 4 doses followed by 1,000 mg intravenously every 6 weeks.
The main efficacy endpoints were overall response rate (ORR) and duration of response (DOR), as assessed by blinded independent central review (BICR) according to RECIST 1.1. Confirmed ORR was 42.3% (95% CI: 30.6%, 54.6%). The complete response rate was 12.7% and partial response rate was 29.6%. Median DOR was not reached, with 93.3% of patients having durations ≥6 months (range: 2.6 to 22.4 months, ongoing at last assessment).
Serious adverse reactions occurred in 34% of patients receiving dostarlimab-gxly. Serious adverse reactions in >2% of patients included sepsis , acute kidney injury , urinary tract infection , abdominal pain , and pyrexia . The most common adverse reactions (≥20%) were fatigue/asthenia, nausea, diarrhea, anemia, and constipation. The most common grade 3 or 4 adverse reactions (≥2%) were anemia and transaminases increased. Immune-mediated adverse reactions can occur including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis.
The recommended dostarlimab-gxly dose and schedule (doses 1 through 4) is 500 mg every 3 weeks. Subsequent dosing, beginning 3 weeks after dose 4, is 1,000 mg every 6 weeks until disease progression or unacceptable toxicity. Dostarlimab-gxly should be administered as an intravenous infusion over 30 minutes.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
FDA also approved the VENTANA MMR RxDx Panel as a companion diagnostic device for selecting endometrial cancer patients for treatment with dostarlimab-gxly.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
Serious adverse reactions in >2% of patients included sepsis, acute kidney injury, urinary tract infection, abdominal pain, and pyrexia.[1][2]
Immune-mediated adverse reactions can occur including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis.[1][2]
History
Like several other available and experimental monoclonal antibodies, it is a PD-1 inhibitor. As of 2020, it is undergoing Phase I/II and Phase III clinical trials.[6][7][8] The manufacturer, Tesaro, announced prelimary successful results from the Phase I/II GARNET study.[6][9][10]
In 2020, the GARNET study announced that Dostarlimab was demonstrating potential to treat a subset of women with recurrent or advanced endometrial cancer.[11]
April 2021, Dostarlimab is approved for the treatment of recurrent or advanced endometrial cancer with deficient mismatch repair (dMMR), which are genetic anomalies abnormalities that disrupt DNA repair.[12]
On April 22, 2021, the Food and Drug Administration granted accelerated approval to dostarlimab-gxly (Jemperli, GlaxoSmithKline LLC).[1] Efficacy was evaluated based on cohort (A1) in GARNET Trial (NCT02715284), a multicenter, multicohort, open-label trial in patients with advanced solid tumors.[1]
Society and culture
Legal status
On 25 February 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a conditional marketing authorization for the medicinal product Jemperli, intended for the treatment of certain types of recurrent or advanced endometrial cancer.[13] The applicant for this medicinal product is GlaxoSmithKline (Ireland) Limited.[13]
^ Jump up to:ab Clinical trial number NCT02715284 for “A Phase 1 Dose Escalation and Cohort Expansion Study of TSR-042, an Anti-PD-1 Monoclonal Antibody, in Patients With Advanced Solid Tumors (GARNET)” at ClinicalTrials.gov
^ Clinical trial number NCT03981796 for “A Study of Dostarlimab (TSR-042) Plus Carboplatin-paclitaxel Versus Placebo Plus Carboplatin-paclitaxel in Patients With Recurrent or Primary Advanced Endometrial Cancer (RUBY)” at ClinicalTrials.gov
^ Clinical trial number NCT03602859 for “A Phase 3 Comparison of Platinum-Based Therapy With TSR-042 and Niraparib Versus Standard of Care Platinum-Based Therapy as First-Line Treatment of Stage III or IV Nonmucinous Epithelial Ovarian Cancer (FIRST)” at ClinicalTrials.gov
“Dostarlimab”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT02715284 for “Study of TSR-042, an Anti-programmed Cell Death-1 Receptor (PD-1) Monoclonal Antibody, in Participants With Advanced Solid Tumors (GARNET)” at ClinicalTrials.gov
Kaplon H, Muralidharan M, Schneider Z, Reichert JM: Antibodies to watch in 2020. MAbs. 2020 Jan-Dec;12(1):1703531. doi: 10.1080/19420862.2019.1703531. [Article]
Temrikar ZH, Suryawanshi S, Meibohm B: Pharmacokinetics and Clinical Pharmacology of Monoclonal Antibodies in Pediatric Patients. Paediatr Drugs. 2020 Apr;22(2):199-216. doi: 10.1007/s40272-020-00382-7. [Article]
Green AK, Feinberg J, Makker V: A Review of Immune Checkpoint Blockade Therapy in Endometrial Cancer. Am Soc Clin Oncol Educ Book. 2020 Mar;40:1-7. doi: 10.1200/EDBK_280503. [Article]
Deshpande M, Romanski PA, Rosenwaks Z, Gerhardt J: Gynecological Cancers Caused by Deficient Mismatch Repair and Microsatellite Instability. Cancers (Basel). 2020 Nov 10;12(11). pii: cancers12113319. doi: 10.3390/cancers12113319. [Article]
FDA Approved Drug Products: Jemperli (dostarlimab-gxly) for intravenous injection [Link]
FDA News Release: FDA grants accelerated approval to dostarlimab-gxly for dMMR endometrial cancer [Link]
Statement on a Nonproprietary Name Adopted by the USAN Council: Dostarlimab [Link]
Loncastuximab tesirine-lpyl is a CD19-directed antibody and alkylating agent conjugate, consisting of a humanized IgG1 kappa monoclonal antibody conjugated to SG3199, a pyrrolobenzodiazepine (PBD) dimer cytotoxic alkylating agent, through a protease-cleavable valine–alanine linker. SG3199 attached to the linker is designated as SG3249, also known as tesirine.
Loncastuximab tesirine-lpyl has an approximate molecular weight of 151 kDa. An average of 2.3 molecules of SG3249 are attached to each antibody molecule. Loncastuximab tesirine-lpyl is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells, and the small molecule components are produced by chemical synthesis.
ZYNLONTA (loncastuximab tesirine-lpyl) for injection is supplied as a sterile, white to off-white, preservative-free, lyophilized powder, which has a cake-like appearance, for intravenous infusion after reconstitution and dilution. Each single-dose vial delivers 10 mg of loncastuximab tesirine-lpyl, L-histidine (2.8 mg), L-histidine monohydrochloride (4.6 mg), polysorbate 20 (0.4 mg), and sucrose (119.8 mg). After reconstitution with 2.2 mL Sterile Water for Injection, USP, the final concentration is 5 mg/mL with a pH of approximately 6.0.
Loncastuximab tesirine , sold under the brand name Zynlonta, is used for the treatment of large B-cell lymphoma. It is an antibody-drug conjugate (ADC) composed of a humanized antibody targeting the protein CD19, which is expressed in a wide range of B cell hematological tumors.[2] The experimental drug, developed by ADC Therapeutics is being tested in clinical trials for the treatment of B-cell non-Hodgkin lymphoma (NHL) and B-cell acute lymphoblastic leukemia (ALL).
On April 23, 2021, the Food and Drug Administration granted accelerated approval to loncastuximab tesirine-lpyl (Zynlonta, ADC Therapeutics SA), a CD19-directed antibody and alkylating agent conjugate, for adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, DLBCL arising from low grade lymphoma, and high-grade B-cell lymphoma.
Approval was based on LOTIS-2 (NCT03589469), an open-label, single-arm trial in 145 adult patients with relapsed or refractory DLBCL or high-grade B-cell lymphoma after at least two prior systemic regimens. Patients received loncastuximab tesirine-lpyl 0.15 mg/kg every 3 weeks for 2 cycles, then 0.075 mg/kg every 3 weeks for subsequent cycles. Patients received treatment until progressive disease or unacceptable toxicity.
The main efficacy outcome measure was overall response rate (ORR), as assessed by an independent review committee using Lugano 2014 criteria. The ORR was 48.3% (95% CI: 39.9, 56.7) with a complete response rate of 24.1% (95% CI: 17.4, 31.9). After a median follow-up of 7.3 months, median response duration was 10.3 months (95% CI: 6.9, NE). Of the 70 patients who achieved objective responses, 36% were censored for response duration prior to 3 months.
Most common (≥20%) adverse reactions in patients receiving loncastuximab tesirine-lpyl, including laboratory abnormalities, are thrombocytopenia, increased gamma-glutamyltransferase, neutropenia, anemia, hyperglycemia, transaminase elevation, fatigue, hypoalbuminemia, rash, edema, nausea, and musculoskeletal pain.
The prescribing information provides warnings and precautions for adverse reactions including edema and effusions, myelosuppression, infections, and cutaneous reactions.
The recommended loncastuximab tesirine-lpyl dosage is 0.15 mg/kg every 3 weeks for 2 cycles, then 0.075 mg/kg every 3 weeks for subsequent cycles, by intravenous infusion over 30 minutes on day 1 of each cycle (every 3 weeks). Patients should be premedicated with dexamethasone 4 mg orally or intravenously twice daily for 3 days beginning the day before loncastuximab tesirine-lpyl.
Technology
The humanized monoclonal antibody is stochastically conjugated via a valine-alanine cleavable, maleimide linker to a cytotoxic (anticancer) pyrrolobenzodiazepine (PBD) dimer. The antibody binds to CD19, a protein which is highly expressed on the surface of B-cell hematological tumors[3] including certain forms of lymphomas and leukemias. After binding to the tumor cells the antibody is internalized, the cytotoxic drug PBD is released and the cancer cells are killed. PBD dimers are generated out of PBD monomers, a class of natural products produced by various actinomycetes. PBD dimers work by crosslinking specific sites of the DNA, blocking the cancer cells’ division that cause the cells to die. As a class of DNA-crosslinking agents they are significantly more potent than systemic chemotherapeutic drugs.[4]
Clinical trials
Two phase I trials are evaluating the drug in patients with relapsed or refractory B-cell non-Hodgkin’s lymphoma and relapsed or refractory B-cell acute lymphoblastic leukemia.[5] At the 14th International Conference on Malignant Lymphoma interim results from a Phase I, open-label, dose-escalating study designed to evaluate the treatment of loncastuximab tesirine in relapsed or refractory non-Hodgkin’s lymphoma were presented.[6] Among the patients enrolled at the time of the data cutoff the overall response rate was 61% in the total patient population (42% complete response and 19% partial response) and in patients with relapsing or refractory diffuse large B-cell lymphoma (DLBCL) the overall response rate was 57% (43% complete response and 14% partial response).[7][8]
Orphan drug designation
Loncastuximab tesirine was granted Orphan Drug Designation by the U.S. Food and Drug Administration (FDA) for the treatment of diffuse large B-cell lymphoma and mantle cell lymphoma.[9]
Sotorasib is an inhibitor of the RAS GTPase family. The molecular formula is C30H30F2N6O3, and the molecular weight is 560.6 g/mol. The chemical name of sotorasib is 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2enoyl) piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one. The chemical structure of sotorasib is shown below:
Sotorasib has pKa values of 8.06 and 4.56. The solubility of sotorasib in the aqueous media decreases over the range pH 1.2 to 6.8 from 1.3 mg/mL to 0.03 mg/mL.
LUMAKRAS is supplied as film-coated tablets for oral use containing 120 mg of sotorasib. Inactive ingredients in the tablet core are microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, and magnesium stearate. The film coating material consists of polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide yellow.
FDA grants accelerated approval to sotorasib for KRAS G12C mutated NSCLC
On May 28, 2021, the Food and Drug Administration granted accelerated approval to sotorasib (Lumakras, Amgen, Inc.), a RAS GTPase family inhibitor, for adult patients with KRAS G12C ‑mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA ‑approved test, who have received at least one prior systemic therapy.
FDA also approved the QIAGEN therascreen® KRAS RGQ PCR kit (tissue) and the Guardant360® CDx (plasma) as companion diagnostics for Lumakras. If no mutation is detected in a plasma specimen, the tumor tissue should be tested.
Approval was based on CodeBreaK 100, a multicenter, single-arm, open label clinical trial (NCT03600883) which included patients with locally advanced or metastatic NSCLC with KRAS G12C mutations. Efficacy was evaluated in 124 patients whose disease had progressed on or after at least one prior systemic therapy. Patients received sotorasib 960 mg orally daily until disease progression or unacceptable toxicity.
The main efficacy outcome measures were objective response rate (ORR) according to RECIST 1.1, as evaluated by blinded independent central review and response duration. The ORR was 36% (95% CI: 28%, 45%) with a median response duration of 10 months (range 1.3+, 11.1).
The most common adverse reactions (≥ 20%) were diarrhea, musculoskeletal pain, nausea, fatigue, hepatotoxicity, and cough. The most common laboratory abnormalities (≥ 25%) were decreased lymphocytes, decreased hemoglobin, increased aspartate aminotransferase, increased alanine aminotransferase, decreased calcium, increased alkaline phosphatase, increased urine protein, and decreased sodium.
The recommended sotorasib dose is 960 mg orally once daily with or without food.
The approved 960 mg dose is based on available clinical data, as well as pharmacokinetic and pharmacodynamic modeling that support the approved dose. As part of the evaluation for this accelerated approval, FDA is requiring a postmarketing trial to investigate whether a lower dose will have a similar clinical effect.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada, and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, the Assessment Aid, and the Product Quality Assessment Aid (PQAA), voluntary submissions from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 10 weeks ahead of the FDA goal date.
The most common side effects include diarrhea, musculoskeletal pain, nausea, fatigue, liver damage and cough.[1][2]
Sotorasib is an inhibitor of the RAS GTPase family.[1]
Sotorasib is the first approved targeted therapy for tumors with any KRAS mutation, which accounts for approximately 25% of mutations in non-small cell lung cancers.[2] KRAS G12C mutations represent about 13% of mutations in non-small cell lung cancers.[2] Sotorasib was approved for medical use in the United States in May 2021.[2][5]
Sotorasib is an experimental KRAS inhibitor being investigated for the treatment of KRAS G12C mutant non small cell lung cancer, colorectal cancer, and appendix cancer.
Sotorasib, also known as AMG-510, is an acrylamide derived KRAS inhibitor developed by Amgen.1,3 It is indicated in the treatment of adult patients with KRAS G12C mutant non small cell lung cancer.6 This mutation makes up >50% of all KRAS mutations.2 Mutant KRAS discovered in 1982 but was not considered a druggable target until the mid-2010s.5 It is the first experimental KRAS inhibitor.1
The drug MRTX849 is also currently being developed and has the same target.1
Sotorasib was granted FDA approval on 28 May 2021.6
Medical uses
Sotorasib is indicated for the treatment of adults with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.[1][2]
Clinical development
Sotorasib is being developed by Amgen. Phase I clinical trials were completed in 2020.[6][7][8] In December 2019, it was approved to begin Phase II clinical trials.[9]
Because the G12C KRAS mutation is relatively common in some cancer types, 14% of non-small-cell lung cancer adenocarcinoma patients and 5% of colorectal cancer patients,[10] and sotorasib is the first drug candidate to target this mutation, there have been high expectations for the drug.[10][11][12] The Food and Drug Administration has granted a fast track designation to sotorasib for the treatment of metastatic non-small-cell lung carcinoma with the G12C KRAS mutation.[13]
Chemistry and pharmacology
Sotorasib can exist in either of two atropisomeric forms and one is more active than the other.[10] It selectively forms an irreversible covalent bond to the sulfur atom in the cysteine residue that is present in the mutated form of KRAS, but not in the normal form.[10]
History
Researchers evaluated the efficacy of sotorasib in a study of 124 participants with locally advanced or metastatic KRAS G12C-mutated non-small cell lung cancer with disease progression after receiving an immune checkpoint inhibitor and/or platinum-based chemotherapy.[2] The major outcomes measured were objective response rate (proportion of participants whose tumor is destroyed or reduced) and duration of response.[2] The objective response rate was 36% and 58% of those participants had a duration of response of six months or longer.[2]
KRAS is the most frequently mutated oncogene in cancer and encodes a key signalling protein in tumours1,2. The KRAS(G12C) mutant has a cysteine residue that has been exploited to design covalent inhibitors that have promising preclinical activity3,4,5. Here we optimized a series of inhibitors, using novel binding interactions to markedly enhance their potency and selectivity. Our efforts have led to the discovery of AMG 510, which is, to our knowledge, the first KRAS(G12C) inhibitor in clinical development. In preclinical analyses, treatment with AMG 510 led to the regression of KRASG12C tumours and improved the anti-tumour efficacy of chemotherapy and targeted agents. In immune-competent mice, treatment with AMG 510 resulted in a pro-inflammatory tumour microenvironment and produced durable cures alone as well as in combination with immune-checkpoint inhibitors. Cured mice rejected the growth of isogenic KRASG12D tumours, which suggests adaptive immunity against shared antigens. Furthermore, in clinical trials, AMG 510 demonstrated anti-tumour activity in the first dosing cohorts and represents a potentially transformative therapy for patients for whom effective treatments are lacking.
Paper
Scientific Reports (2020), 10(1), 11992
PAPER
European journal of medicinal chemistry (2021), 213, 113082.
KRAS is the most commonly altered oncogene of the RAS family, especially the G12C mutant (KRASG12C), which has been a promising drug target for many cancers. On the basis of the bicyclic pyridopyrimidinone framework of the first-in-class clinical KRASG12C inhibitor AMG510, a scaffold hopping strategy was conducted including a F–OH cyclization approach and a pyridinyl N-atom working approach leading to new tetracyclic and bicyclic analogues. Compound 26a was identified possessing binding potency of 1.87 μM against KRASG12C and cell growth inhibition of 0.79 μM in MIA PaCa-2 pancreatic cancer cells. Treatment of 26a with NCI–H358 cells resulted in down-regulation of KRAS-GTP levels and reduction of phosphorylation of downstream ERK and AKT dose-dependently. Molecular docking suggested that the fluorophenol moiety of 26a occupies a hydrophobic pocket region thus forming hydrogen bonding to Arg68. These results will be useful to guide further structural modification.
PAPER
Journal of Medicinal Chemistry (2020), 63(1), 52-65.
KRASG12C has emerged as a promising target in the treatment of solid tumors. Covalent inhibitors targeting the mutant cysteine-12 residue have been shown to disrupt signaling by this long-“undruggable” target; however clinically viable inhibitors have yet to be identified. Here, we report efforts to exploit a cryptic pocket (H95/Y96/Q99) we identified in KRASG12C to identify inhibitors suitable for clinical development. Structure-based design efforts leading to the identification of a novel quinazolinone scaffold are described, along with optimization efforts that overcame a configurational stability issue arising from restricted rotation about an axially chiral biaryl bond. Biopharmaceutical optimization of the resulting leads culminated in the identification of AMG 510, a highly potent, selective, and well-tolerated KRASG12C inhibitor currently in phase I clinical trials (NCT03600883).
The present disclosure relates to an improved, efficient, scalable process to prepare intermediate compounds, such as compound of Formula 6A, having the structure,
useful for the synthesis of compounds for the treatment of KRAS G12C mutated cancers.
BACKGROUND
[0003] KRAS gene mutations are common in pancreatic cancer, lung adenocarcinoma, colorectal cancer, gall bladder cancer, thyroid cancer, and bile duct cancer. KRAS mutations are also observed in about 25% of patients with NSCLC, and some studies have indicated that KRAS mutations are a negative prognostic factor in patients with NSCLC. Recently, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations have been found to confer resistance to epidermal growth factor receptor (EGFR) targeted therapies in colorectal cancer; accordingly, the mutational status of KRAS can provide important information prior to the prescription of TKI therapy. Taken together, there is a need for new medical treatments for patients with pancreatic cancer, lung adenocarcinoma, or colorectal cancer, especially those who have been diagnosed to have such cancers characterized by a KRAS mutation, and including those who have progressed after chemotherapy.
Related Synthetic Processes
[0126] The following intermediate compounds of 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one are representative examples of the disclosure and are not intended to be construed as limiting the scope of the present invention.
[0127] A synthesis of Compound 9 and the relevant intermediates is described in U.S. Serial No.15/984,855, filed May 21, 2018 (U.S. Publication No.2018/0334454, November 22, 2018) which claims priority to and the benefit claims the benefit of U.S. Provisional Application No.62/509,629, filed on May 22, 2017, both of which are incorporated herein by reference in their entireties for all purposes. 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one was prepared using the following process, in which the isomers of the final product were isolated via chiral chromatography.
[0128] Step 1: 2,6-Dichloro-5-fluoronicotinamide (Intermediate S). To a mixture of 2,6-dichloro-5-fluoro-nicotinic acid (4.0 g, 19.1 mmol, AstaTech Inc., Bristol, PA) in dichloromethane (48 mL) was added oxalyl chloride (2M solution in DCM, 11.9 mL, 23.8 mmol), followed by a catalytic amount of DMF (0.05 mL). The reaction was stirred at room temperature overnight and then was concentrated. The residue was dissolved in 1,4-dioxane (48 mL) and cooled to 0 °C. Ammonium hydroxide solution (28.0-30% NH3 basis, 3.6 mL, 28.6 mmol) was added slowly via syringe. The resulting mixture was stirred at 0 °C for 30 min and then was concentrated. The residue was diluted with a 1:1 mixture of EtOAc/Heptane and agitated for 5 min, then was filtered. The filtered solids were discarded, and the remaining mother liquor was partially concentrated to half volume and filtered. The filtered solids were washed with heptane and dried in a reduced-pressure oven (45 °C) overnight to provide 2,6-dichloro-5-fluoronicotinamide. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.23 (d, J = 7.9 Hz, 1 H) 8.09 (br s, 1 H) 7.93 (br s, 1 H). m/z (ESI, +ve ion): 210.9 (M+H)+.
[0129] Step 2: 2,6-Dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. To an ice-cooled slurry of 2,6-dichloro-5-fluoronicotinamide (Intermediate S, 5.0 g, 23.9 mmol) in THF (20 mL) was added oxalyl chloride (2 M solution in DCM, 14.4 mL, 28.8 mmol) slowly via syringe. The resulting mixture was heated at 75 °C for 1 h, then heating was stopped, and the reaction was concentrated to half volume. After cooling to 0 °C, THF (20 mL) was added, followed by a solution of 2-isopropyl-4-methylpyridin-3-amine (Intermediate R, 3.59 g, 23.92 mmol) in THF (10 mL), dropwise via cannula. The resulting mixture was stirred at 0 °C for 1 h and then was quenched with a 1:1 mixture of brine and saturated aqueous ammonium chloride. The mixture was extracted with EtOAc (3x) and the combined organic layers were dried over anhydrous sodium sulfate and concentrated to provide 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. This material was used without further purification in the following step. m/z (ESI, +ve ion): 385.1(M+H)+.
[0130] Step 3: 7-Chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione. To an ice-cooled solution of 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (9.2 g, 24.0 mmol) in THF (40 mL) was added KHMDS (1 M solution in THF, 50.2 mL, 50.2 mmol) slowly via syringe. The ice bath was removed and the resulting mixture was stirred for 40 min at room temperature. The reaction was quenched with saturated aqueous ammonium chloride and extracted with EtOAc (3x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% 3:1 EtOAc-EtOH/heptane) to provide 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione.1H NMR (400 MHz, DMSO-d6) δ ppm 12.27 (br s, 1H), 8.48-8.55 (m, 2 H), 7.29 (d, J = 4.8 Hz, 1 H), 2.87 (quin, J = 6.6 Hz, 1 H), 1.99-2.06 (m, 3 H), 1.09 (d, J = 6.6 Hz, 3 H), 1.01 (d, J = 6.6 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -126.90 (s, 1 F). m/z (ESI, +ve ion): 349.1 (M+H)+.
[0131] Step 4: 4,7-Dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. To a solution of 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (4.7 g, 13.5 mmol) and DIPEA (3.5 mL, 20.2 mmol) in acetonitrile (20 mL) was added phosphorus oxychloride (1.63 mL, 17.5 mmol), dropwise via syringe. The resulting mixture was heated at 80 °C for 1 h, and then was cooled to room temperature and concentrated to provide 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. This material was used without further purification in the following step. m/z (ESI, +ve ion): 367.1 (M+H)+.
[0132] Step 5: (S)-tert-Butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. To an ice-cooled solution of 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one (13.5 mmol) in acetonitrile (20 mL) was added DIPEA (7.1 mL, 40.3 mmol), followed by (S)-4-N-Boc-2-methyl piperazine (3.23 g, 16.1 mmol, Combi-Blocks, Inc., San Diego, CA, USA). The resulting mixture was warmed to room temperature and stirred for 1 h, then was diluted with cold saturated aqueous sodium bicarbonate solution (200 mL) and EtOAc (300 mL). The mixture was stirred for an additional 5 min, the layers were separated, and the aqueous layer was extracted with more EtOAc (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% EtOAc/heptane) to provide (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. m/z (ESI, +ve ion): 531.2 (M+H)+.
[0133] Step 6: (3S)-tert-Butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. A mixture of (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (4.3 g, 8.1 mmol), potassium trifluoro(2-fluoro-6-hydroxyphenyl)borate (Intermediate Q, 2.9 g, 10.5 mmol), potassium acetate (3.2 g, 32.4 mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane (661 mg, 0.81 mmol) in 1,4-dioxane (80 mL) was degassed with nitrogen for 1 min. De-oxygenated water (14 mL) was added, and the resulting mixture was heated at 90 °C for 1 h. The reaction was allowed to cool to room temperature, quenched with half-saturated aqueous sodium bicarbonate, and extracted with EtOAc (2x) and DCM (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-60% 3:1 EtOAc-EtOH/heptane) to provide (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate.1H NMR (400 MHz, DMSO-d6) δ ppm 10.19 (br s, 1 H), 8.38 (d, J = 5.0 Hz, 1 H), 8.26 (dd, J = 12.5, 9.2 Hz, 1 H), 7.23-7.28 (m, 1 H), 7.18 (d, J = 5.0 Hz, 1 H), 6.72 (d, J = 8.0 Hz, 1 H), 6.68 (t, J = 8.9 Hz, 1 H), 4.77-4.98 (m, 1 H), 4.24 (br t, J = 14.2 Hz, 1 H), 3.93-4.08 (m, 1 H), 3.84 (br d, J=12.9 Hz, 1 H), 3.52-3.75 (m, 1 H), 3.07-3.28 (m, 1 H), 2.62-2.74 (m, 1 H), 1.86-1.93 (m, 3 H), 1.43-1.48 (m, 9 H), 1.35 (dd, J = 10.8, 6.8 Hz, 3 H), 1.26-1.32 (m, 1 H), 1.07 (dd, J = 6.6, 1.7 Hz, 3 H), 0.93 (dd, J = 6.6, 2.1 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -115.65 (s, 1 F), -128.62 (s, 1 F). m/z (ESI, +ve ion): 607.3 (M+H)+.
[0134] Step 7: 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one. Trifluoroacetic acid (25 mL, 324 mmol) was added to a solution of (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (6.3 g, 10.4 mmol) in DCM (30 mL). The resulting mixture was stirred at room temperature for 1 h and then was concentrated. The residue was dissolved in DCM (30 mL), cooled to 0 °C, and sequentially treated with DIPEA (7.3 mL, 41.7 mmol) and a solution of acryloyl chloride (0.849 mL, 10.4 mmol) in DCM (3 mL; added dropwise via syringe). The reaction was stirred at 0 °C for 10 min, then was quenched with half-saturated aqueous sodium bicarbonate and extracted with DCM (2x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-100% 3:1 EtOAc-EtOH/heptane) to provide 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one.1H NMR (400 MHz, DMSO-d6) δ ppm 10.20 (s, 1 H), 8.39 (d, J = 4.8 Hz, 1 H), 8.24-8.34 (m, 1 H), 7.23-7.32 (m, 1 H), 7.19 (d, J = 5.0 Hz, 1 H), 6.87 (td, J = 16.3, 11.0 Hz, 1 H), 6.74 (d, J = 8.6 Hz, 1 H), 6.69 (t, J = 8.6 Hz, 1 H), 6.21 (br d, J = 16.2 Hz, 1 H), 5.74-5.80 (m, 1 H), 4.91 (br s, 1 H), 4.23-4.45 (m, 2 H), 3.97-4.21 (m, 1 H), 3.44-3.79 (m, 2 H), 3.11-3.31 (m, 1 H), 2.67-2.77 (m, 1 H), 1.91 (s, 3 H), 1.35 (d, J = 6.8 Hz, 3 H), 1.08 (d, J = 6.6 Hz, 3 H), 0.94 (d, J = 6.8 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ ppm -115.64 (s, 1 F), -128.63 (s, 1 F). m/z (ESI, +ve ion): 561.2 (M+H)+.
[0135] Another synthesis of Compound 9 and the relevant intermediates was described in a U.S. provisional patent application filed November 16, 2018, which is incorporated herein by reference in its entirety for all purposes.
Representative Synthetic Processes
[0136] The present disclosure comprises the following steps wherein the synthesis and utilization of the boroxine intermediate is a novel and inventive step in the manufacture of AMG 510 (Compound 9):
Raw Materials
Step la
[0137] To a solution of 2,6-dichloro-5-fluoro-3-pyridinecarboxylic acid (25kg; 119. lmol) in dichloromethane (167kg) and DMF (592g) was added Oxalyl chloride (18.9kg; 148.9mol) while maintaining an internal temp between 15-20 °C. Additional dichloromethane (33kg) was added as a rinse and the reaction mixture stirred for 2h. The reaction mixture is cooled then quenched with ammonium hydroxide (40.2L; 595.5mol) while maintaining internal temperature 0 ± 10°C. The resulting slurry was stirred for 90min then the product collected by filtration. The filtered solids were washed with DI water (3X 87L) and dried to provide 2,6-dichloro-5-fluoronicotinamide (Compound 1).
Step 1b
[0138] In reactor A, a solution of 2,6-dichloro-5-fluoronicotinamide (Compound 1) (16.27kg; 77.8mol) in dichloromethane (359.5kg) was added oxalyl chloride (11.9kg;
93.8mol) while maintaining temp ≤ 25°C for 75min. The resulting solution was then headed to 40°C ± 3°C and aged for 3h. Using vacuum, the solution was distilled to remove dichloromethane until the solution was below the agitator. Dichloromethane (300 kg) was then added and the mixture cooled to 0 ± 5°C. To a clean, dry reactor (reactor B) was added,2-isopropyl-4-methylpyridin-3-amine (ANILINE Compound 2A) (12.9kg; 85.9mol) followed by dichloromethane (102.6 kg). The ANILINE solution was azeodried via vacuum distillation while maintaining an internal temperature between 20-25 °), replacing with additional dichloromethane until the solution was dry by KF analysis (limit ≤ 0.05%). The solution volume was adjusted to approx. 23L volume with dichloromethane. The dried ANILINE solution was then added to reactor A while maintaining an internal temperature of 0 ± 5°C throughout the addition. The mixture was then heated to 23 °C and aged for 1h. the solution was polish filtered into a clean reactor to afford 2,6-dichloro-5-fluoro-N-((2- isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (Compound 3) as a solution in DCM and used directly in the next step.
Step 2
[0139] A dichloromethane solution of 2,6-dichloro-5-fluoro-N-{[4-methyl-2-(propan-2- yl)pyridin-3-yl]carbamoyl}pyridine-3-carboxamide (UREA (Compound 3)) (15kg contained; 38.9mol) was solvent exchanged into 2-MeTHF using vacuum distillation while maintaining internal temperature of 20-25 °C. The reactor volume was adjusted to 40L and then
additional 2-MeTHF was charged (105.4 kg). Sodium t-butoxide was added (9.4 kg;
97.8mol) while maintaining 5-10 °C. The contents where warmed to 23 °C and stirred for 3h. The contents where then cooled to 0-5C and ammonium chloride added (23.0kg; 430mol) as a solution in 60L of DI water. The mixture was warmed to 20 C and DI water added (15L) and further aged for 30min. Agitation was stopped and the layers separated. The aqueous layer was removed and to the organic layer was added DI water(81.7L). A mixture of conc HCl (1.5kg) and water (9L) was prepared then added to the reactor slowly until pH measured between 4-5. The layers were separated, and the aqueous layer back extracted using 2-MeTHF (42.2kg). The two organic layers combined and washed with a 10% citric acid solution (75kg) followed by a mixture of water (81.7L) and saturated NaCl (19.8 kg). The organic layer was then washed with saturated sodium bicarbonate (75kg) repeating if necessary to achieve a target pH of ≥ 7.0 of the aqueous. The organic layer was washed again with brine (54.7kg) and then dried over magnesium sulfate (5kg). The mixture was filtered to remove magnesium sulfate rinsing the filtered bed with 2-MeTHF (49.2 kg). The combined filtrate and washes where distilled using vacuum to 40L volume. The concentrated solution was heated to 55 °C and heptane (10-12kg) slowly added until cloud point. The solution was cooled to 23 °C over 2h then heptane (27.3 kg) was added over 2h. The product slurry was aged for 3h at 20-25 °C then filtered and washed with a mixture of 2-MeTHF (2.8kg) and heptane (9kg). The product was dried using nitrogen and vacuum to afford solid 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (rac-DIONE (Compound 4)).
Step 3
[0140] To a vessel, an agitated suspension of Compound 4, (1.0 eq.) in 2- methylterahydrofuran (7.0 L/kg) was added (+)-2,3-dibenzoyl-D-tartaric acid (2.0 eq.) under an atmosphere of nitrogen. 2-MeTHF is chiral, but it is used as a racemic mixture. The different enantiomers of 2-MeTHF are incorporated randomly into the co-crystal. The resulting suspension was warmed to 75°C and aged at 75°C until full dissolution was observed (< 30 mins.). The resulting solution was polish filtered at 75°C into a secondary vessel. To the polish filtered solution was charged n-Heptane (2.0 L/kg) at a rate that maintained the internal temperature above 65°C. The solution was then cooled to 60°C, seeded with crystals (0.01 kg/kg) and allowed to age for 30 minutes. The resulting suspension was cooled to 20°C over 4 hours and then sampled for chiral purity analysis by HPLC. To the suspension, n-Heptane (3.0 L/kg) was charged and then aged for 4 hours at 20°C under an atmosphere of nitrogen. The suspension was filtered, and the isolated solids were washed two times with (2:1) n-Heptane:2-methyltetrahydrofuran (3.0 L/kg). The material was dried with nitrogen and vacuum to afford M-Dione:DBTA: Me-THF complex (Compound 4a).
Step 4
[0141] To vessel A, a suspension of disodium hydrogen phosphate (21.1 kg, 2.0 equiv) in DI water (296.8 L, 6.3 L/kg) was agitated until dissolution was observed (≥ 30 min.). To vessel B, a suspension of the M-Dione:DBTA: Me-THF complex (Composition 4a)[46.9 kg (25.9 kg corrected for M-dione, 1.0 equiv.)] in methyl tert-butyl ether (517.8 L, 11.0 L/kg) was agitated for 15 to 30 minutes. The resulting solution from vessel A was added to vessel B, and then the mixture was agitated for more than 3 hours. The agitation was stopped, and the biphasic mixture was left to separate for more than 30 minutes. The lower aqueous phase was removed and then back extracted with methyl tert-butyl ether (77.7 L, 1.7 L/kg). The organic phases were combined in vessel B and dried with magnesium sulfate (24.8 kg, 0.529 kg/kg). The resulting suspension from vessel B was agitated for more than three hours and then filtered into vessel C. To vessel B, a methyl tert-butyl ether (46.9 L, 1.0 L/kg) rinse was charged and then filtered into vessel C. The contents of vessel C were cooled to 10 °C and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until 320-350 kg (6.8-7.5 kg/kg) of methyl tert-butyl ether was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged a second time over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (195.9 L, 4.2 L/kg) was charged a third time over one hour and then sampled for solvent composition by GC analysis. The vessel C suspension continued to agitate for more than one hour. The suspension was filtered, and then washed with a n-Heptane (68.6 L, 1.5 L/kg) rinse from vessel C. The isolated solids were dried at 50°C, and a sample was submitted for stock suitability. Afforded 7-chloro-6-fluoro-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (M-DIONE) Compound 5M.
[0142] The first-generation process highlighted above has been successfully scaled on 200+ kg of rac-dione starting material (Compound 4). In this process, seeding the crystallization with the thermodynamically-stable rac-dione crystal form (which exhibits low solubility) would cause a batch failure. Based on our subsequent studies, we found that increasing the DBTA equivalents and lowering the seed temperature by adjusting heptane
charge schedule improves robustness of the process. The improved process is resistant to the presence of the thermodynamically-stable rac-dione crystal form and promotes successful separation of atropisomers. Subsequent batches will incorporate the improved process for large scale manufacture.
Step 5
Note: All L/kg amounts are relative to M-Dione input; All equiv. amounts are relative to M-Dione input after adjusted by potency.
[0143] M-Dione (Compound 5M, 1.0 equiv.) and Toluene-1 (10.0 L/kg) was charged to Vessel A. The resulting solution was dried by azeotropic distillation under vacuum at 45 °C until 5.0 L/kg of solvents has been removed. The contents of Vessel A were then cooled to 20 °C.
[0144] Vessel C was charged with Toluene-3 (4.5 L/kg), Phosphoryl chloride (1.5 equiv.) and N,N-Diisopropylethylamine-1 (2.0 equiv.) while maintaining the internal temperature below 20 ± 5 °C.
Upon finishing charging, Vessel C was warmed to 30 ± 5 °C. The contents of Vessel A were then transferred to Vessel C over 4 hours while maintaining the internal temperature at 30 ± 5°C. Vessel A was rinsed with Toluene-2 (0.5 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated at 30°C for an additional 3 hours. The contents of Vessel C were cooled to 20 ± 5 °C. A solution of (s)-1-boc-3-methylpiperazine (1.2 equiv.), N,N-Diisopropylethylamine-2 (1.2 equiv.) in isopropyl acetate-1 (1.0 L/kg) was prepared in Vessel D. The solution of Vessel D was charged to vessel C while maintaining a batch temperature of 20 ± 5 °C (Note: Exotherm is observed). Upon the end of transfer, Vessel D was rinsed with additional dichloromethane (1.0 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated for an additional 60 minutes at 20 °C. A solution of sodium bicarbonate [water-1 (15.0 L/kg + Sodium bicarbonate (4.5 equiv.)] was then charged into Vessel C over an hour while maintaining an internal temperature at 20 ± 5 °C throughout the addition. The contents of Vessel C were agitated for at least 12 hours at which point the Pipazoline (Compound 6) product was isolated by filtration in an agitated filter dryer. The cake was washed with water-2 and -3 (5.0 L/kg x 2 times, agitating each wash for 15 minutes) and isopropyl acetate-2 and 3 (5.0 L/kg x 2 times, agitating each wash for 15 min). The cake as dried under nitrogen for 12 hours.
Acetone Re-slurry (Optional):
[0145] Pipazoline (Compound 6) and acetone (10.0 L/kg) were charged to Vessel E. The suspension was heated to 50 °C for 2 hours. Water-4 (10.0 L/kg) was charged into Vessel E over 1 hour. Upon completion of water addition, the mixture was cooled to 20 °C over 1 hour. The contents of Vessel E were filtered to isolate the product, washing the cake with 1:1 acetone/water mixture (5.0 L/kg). The cake was dried under nitrogen for 12 hours.
Step 6
General Note: All equivalents and volumes are reported in reference to Pipazoline input
Note: All L/kg and kg/kg amounts are relative to Pipazoline input
[0146] Reactor A is charged with Pipazoline (Compound 6, 1.0 equiv), degassed 2- MeTHF (9.0 L/kg) and a solution of potassium acetate (2.0 equiv) in degassed water (6.5 L/kg). The resulting mixture is warmed to 75 ± 5 °C and then, charge a slurry of
Pd(dpePhos)Cl2 (0.003 equiv) in 2-MeTHF (0.5 L/kg). Within 2 h of catalyst charge, a solution of freshly prepared Boroxine (Compound 6A, 0.5 equiv) in wet degassed 2-MeTHF (4.0 L/kg, KF > 4.0%) is charged over the course of >1 hour, but < 2 hours, rinsing with an additional portion of wet 2-MeTHF (0.5 L/kg) after addition is complete. After reaction completion ( <0.15 area % Pipazoline remaining, typically <1 h after boroxine addition is complete), 0.2 wt% (0.002 kg/kg) of Biaryl seed is added as a slurry in 0.02 L/kg wet 2- MeTHF, and the resulting seed bed is aged for > 60 min. Heptane (5.0 L/kg) is added over 2 hours at 75 ± 5 °C. The batch is then cooled to 20 ± 5 °C over 2 hours and aged for an additional 2 h. The slurry is then filtered and cake washed with 1 x 5.0L/kg water, 1 x 5.0L/kg 1:1 iPrOH:water followed by 1 x 5.0 L/kg 1:1 iPrOH:heptane (resuspension wash: the cake is resuspended by agitator and allow to set before filtering) . The cake (Biaryl, Compound 7) is then dried under vacuum with a nitrogen sweep.
Note: If the reaction stalls, an additional charge of catalyst and boroxine is required
Step 7 Charcoal Filtration for Pd removal
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to crude Biaryl input
[0147] In a clean Vessel A, charge crude Biaryl (1 equiv) and charge DCM (10 L/kg). Agitate content for > 60 minutes at 22 ± 5 °C, observing dissolution. Pass crude Biaryl from Vessel A, through a bag filter and carbon filters at a flux ≤ 3 L2/min/m and collect filtrate in clean Vessel B. Charge DCM rinse (1 L/kg) to Vessel A, and through carbon filters to collect in vessel B.
[0148] From filtrate in Vessel B, pull a solution sample for IPC Pd content. Sample is concentrated to solid and analyzed by ICP-MS. IPC: Pd ≤ 25 ppm with respect to Biaryl. a. If Pd content is greater than 25 ppm with respect to Biaryl on first or second IPC sample, pass solution through carbon filter a second time at ≤ 3 L2/min/m2, rinsing with 1 L/kg DCM; sample filtrate for IPC.
b. If Pd content remains greater than 25 ppm after third IPC, install and condition fresh carbon discs. Pass Biaryl filtrate through refreshed carbon filter, washing with 1 L/kg DCM. Sample for IPC.
[0149] Distill and refill to appropriate concentration. Prepare for distillation of recovered filtrate by concentrating to ≤ 4 L/kg DCM, and recharge to reach 5.25 ± 0.25 L/kg DCM prior to moving into Step 7 Boc-deprotection reaction.
Step 7
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to Biaryl input
[0150] To Reactor A was added: tert-butyl (3S)-4-{6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl}-3-methylpiperazine-1-carboxylate (Biaryl) (1.0 equiv), dichloromethane (5.0 L/kg), and the TFA (15.0 equiv, 1.9 L/kg) is charged slowly to maintain the internal temperature at 20 ± 5 °C. The reaction was stirred for 4 h at 20 ± 5 °C.
[0151] To Reactor B was added: potassium carbonate (18.0 equiv), water (20.0 L/kg), and NMP (1.0) to form a homogenous solution. While agitating at the maximum acceptable rate for the equipment, the reaction mixture in A was transferred into the potassium carbonate solution in B over 30 minutes (~ 0.24 L/kg/min rate). The mixture was stirred at 20 ± 5 °C for an additional 12 h.
[0152] The resulting slurry was filtered and rinsed with water (2 x 10 L/kg). The wet cake was dried for 24 h to give 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-4-[(2S)-2-methylpiperazin- 1-yl]-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Des- Boc, Compound 8).
Step 8
Note: All L/kg and kg/kg amounts are relative to Des-Boc input
[0153] Des-Boc (Compound 8, 1.0 equiv) and NMP (4.2 L/kg) are charged to Vessel A under nitrogen, charge the TFA (1.0 equiv.) slowly to maintain the Tr <25 °C. The mixture is aged at 25 °C until full dissolution is observed (about 0.5 hour). The solution is then polish filtered through a 0.45 micron filter into Vessel B, washing with a NMP (0.8 L/kg). The filtrate and wash are combined, and then cooled to 0 °C. To the resulting solution, Acryloyl Chloride (1.3 equiv.) is added while maintaining temperature < 10 C. The reaction mixture is then aged at 5 ±5°C until completed by IPC (ca.1.5 hrs).
Preparation of Aqueous Disodium Phosphate Quench:
[0154] Disodium Phosphate (3.0 equiv) and Water (15.0 L/kg) are charged to Vessel C. The mixture is aged at 25 °C until full dissolution is observed. The solution is warmed to 45 ±5°C. A seed slurry of AMG 510 (0.005 equiv.) in Water (0.4 L/kg) is prepared and added to Vessel C while maintaining temperature at 45 ±5°C.
[0155] The reaction mixture in Vessel B is transferred to Vessel C (quench solution) while maintaining temperature at 45 ±5°C (ca.1 hrs). Vessel B is washed with a portion of NMP (0.5 L/kg). The product slurry is aged for 2 hrs at 45 ±5°C, cooled to 20 °C over 3 hrs, aged at 20 °C for a minimum of 12 hrs, filtered and washed with Water (2 x 10.0 L/kg). The product is dried using nitrogen and vacuum to afford Crude AMG 510 (Compound 9A).
Step 9
General Note: All equivalents and volumes are reported in reference to crude AMG 510 input
Note: All L/kg and kg/kg amounts are relative to Crude AMG 510 input
[0156] Reactor A was charged with 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4- methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1- yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Crude AMG 510) (1.0 equiv), ethanol (7.5 L/kg), and water (1.9 L/kg). The mixture heated to 75 °C and polish filtered into a clean Reactor B. The solution was cool to 45 °C and seeded with authentic milled AMG 510 seed (0.015 േ 0.005
1 Seed performs best when reduced in particle size via milling or with other type of mechanical grinding if mill is not available (mortar/ pestle). Actual seed utilized will be based on seed availability. 1.0- 2.0% is seed is target amount.
kg/kg); the resulting slurry was aged for 30 min. Water (15.0 L/kg) was added over 5h while maintaining an internal temperature > 40 °C; the mixture was aged for an additional 2h.
[0157] The mixture was cooled to 20 °C over 3 hours and aged for 8h, after which the solid was collected by filtration and washed using a mixture of ethanol (2.5 L/kg) and water (5.0 L/kg). The solid was dried using vacuum and nitrogen to obtain 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510, Compound 9).
Compound 6A Boroxine Synthesis:
Lithiation/borylation
[0158] Reactor A was charged with THF (6 vol), a secondary amine base, Diisopropylamine (1.4 equiv), and a catalyst, such as triethylamine hydrochloride (0.01 equiv.). The resulting solution was cooled to -70 °C and a first base, n-BuLi (2.5 M in hexane, 1.5 equiv) was slowly added. After addition is complete, a solution of 3-fluoroanisole (1.0 equiv) in THF (6 vol) was added slowly and kept at -70 °C for 5 min. Concurrently or subsequently, a reagent, B(EtO)3 (2.0 equiv), was added slowly and kept at -70 °C for 10 min. The reaction mixture was quenched with an acid, 2N HCl. The quenched reaction mixture was extracted with MTBE (3 x 4 vol). The combined organic phases were concentrated to 1.5-3 total volumes. Heptane (7-9 vol) was added drop-wise and the mixture was cooled to 0-10 °C and stirred for 3 h. The mixture was filtrated and rinsed with heptane (1.5 vol). The solid was dried under nitrogen at < 30 °C to afford (2-fluoro-6-methoxyphenyl)boronic acid.
Demethylation:
Note: All L/kg and kg/kg amounts are relative to (2-fluoro-6-methoxyphenyl)boronic acid input
[0159] To a reactor, charge dichloromethane (solvent, 4.0 L/kg) and an acid, BBr3 (1.2 equiv), and cool to -20 °C. To this solution, a suspension of (2-fluoro-6-methoxyphenyl)boronic acid (1.0 equiv) in dichloromethane (4.0 L/kg) was added into the BBr3/DCM mixture while keeping temperature -15 to -25 °C. The reaction was allowed to proceed for approximately 2 hours while monitored by HPLC [≤1% (2-fluoro-6-methoxyphenyl)boronic acid] before reverse quenching into water (3.0 L/kg). The precipitated solid was then isolated by filtration and slurried with water (3.0 L/kg) on the filter prior to deliquoring. The filtrates were adjusted to pH 4-6 by the addition of sodium bicarbonate. The bottom organic phase was separated and the resulting aqueous layer was washed with dichloromethane (solvent, 5.0 Vol) and adjusted to pH = 1 by addition of concentrated hydrochloric acid. The resulting solids were isolated by filtration, washing the cake with water (2 x 5.0 L/kg)
Purification via Reslurry (required)
[0160] The combined crude solids were charged into a reactor and slurried with 5% EtOH/water (5.0 L/kg) at 20 °C for >1 h. The purified product was then isolated by filtration and rinsed with water (2 x 3 L/kg) before drying on the filter at < 30 °C to with nitrogen/vacuum to afford 2,2′,2”-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tris(3-fluorophenol) (Boroxine, Compound 6A).
^ Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation ” at ClinicalTrials.gov
“Sotorasib”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation (CodeBreaK 100)” at ClinicalTrials.gov
Sotorasib is an inhibitor of the RAS GTPase family. The molecular formula is C30H30F2N6O3, and the molecular weight is 560.6 g/mol. The chemical name of sotorasib is 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2enoyl) piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one. The chemical structure of sotorasib is shown below:
Sotorasib has pKa values of 8.06 and 4.56. The solubility of sotorasib in the aqueous media decreases over the range pH 1.2 to 6.8 from 1.3 mg/mL to 0.03 mg/mL.
LUMAKRAS is supplied as film-coated tablets for oral use containing 120 mg of sotorasib. Inactive ingredients in the tablet core are microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, and magnesium stearate. The film coating material consists of polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide yellow.
FDA grants accelerated approval to sotorasib for KRAS G12C mutated NSCLC
On May 28, 2021, the Food and Drug Administration granted accelerated approval to sotorasib (Lumakras, Amgen, Inc.), a RAS GTPase family inhibitor, for adult patients with KRAS G12C ‑mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA ‑approved test, who have received at least one prior systemic therapy.
FDA also approved the QIAGEN therascreen® KRAS RGQ PCR kit (tissue) and the Guardant360® CDx (plasma) as companion diagnostics for Lumakras. If no mutation is detected in a plasma specimen, the tumor tissue should be tested.
Approval was based on CodeBreaK 100, a multicenter, single-arm, open label clinical trial (NCT03600883) which included patients with locally advanced or metastatic NSCLC with KRAS G12C mutations. Efficacy was evaluated in 124 patients whose disease had progressed on or after at least one prior systemic therapy. Patients received sotorasib 960 mg orally daily until disease progression or unacceptable toxicity.
The main efficacy outcome measures were objective response rate (ORR) according to RECIST 1.1, as evaluated by blinded independent central review and response duration. The ORR was 36% (95% CI: 28%, 45%) with a median response duration of 10 months (range 1.3+, 11.1).
The most common adverse reactions (≥ 20%) were diarrhea, musculoskeletal pain, nausea, fatigue, hepatotoxicity, and cough. The most common laboratory abnormalities (≥ 25%) were decreased lymphocytes, decreased hemoglobin, increased aspartate aminotransferase, increased alanine aminotransferase, decreased calcium, increased alkaline phosphatase, increased urine protein, and decreased sodium.
The recommended sotorasib dose is 960 mg orally once daily with or without food.
The approved 960 mg dose is based on available clinical data, as well as pharmacokinetic and pharmacodynamic modeling that support the approved dose. As part of the evaluation for this accelerated approval, FDA is requiring a postmarketing trial to investigate whether a lower dose will have a similar clinical effect.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada, and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, the Assessment Aid, and the Product Quality Assessment Aid (PQAA), voluntary submissions from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 10 weeks ahead of the FDA goal date.
The most common side effects include diarrhea, musculoskeletal pain, nausea, fatigue, liver damage and cough.[1][2]
Sotorasib is an inhibitor of the RAS GTPase family.[1]
Sotorasib is the first approved targeted therapy for tumors with any KRAS mutation, which accounts for approximately 25% of mutations in non-small cell lung cancers.[2] KRAS G12C mutations represent about 13% of mutations in non-small cell lung cancers.[2] Sotorasib was approved for medical use in the United States in May 2021.[2][5]
Sotorasib is an experimental KRAS inhibitor being investigated for the treatment of KRAS G12C mutant non small cell lung cancer, colorectal cancer, and appendix cancer.
Sotorasib, also known as AMG-510, is an acrylamide derived KRAS inhibitor developed by Amgen.1,3 It is indicated in the treatment of adult patients with KRAS G12C mutant non small cell lung cancer.6 This mutation makes up >50% of all KRAS mutations.2 Mutant KRAS discovered in 1982 but was not considered a druggable target until the mid-2010s.5 It is the first experimental KRAS inhibitor.1
The drug MRTX849 is also currently being developed and has the same target.1
Sotorasib was granted FDA approval on 28 May 2021.6
Medical uses
Sotorasib is indicated for the treatment of adults with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.[1][2]
Clinical development
Sotorasib is being developed by Amgen. Phase I clinical trials were completed in 2020.[6][7][8] In December 2019, it was approved to begin Phase II clinical trials.[9]
Because the G12C KRAS mutation is relatively common in some cancer types, 14% of non-small-cell lung cancer adenocarcinoma patients and 5% of colorectal cancer patients,[10] and sotorasib is the first drug candidate to target this mutation, there have been high expectations for the drug.[10][11][12] The Food and Drug Administration has granted a fast track designation to sotorasib for the treatment of metastatic non-small-cell lung carcinoma with the G12C KRAS mutation.[13]
Chemistry and pharmacology
Sotorasib can exist in either of two atropisomeric forms and one is more active than the other.[10] It selectively forms an irreversible covalent bond to the sulfur atom in the cysteine residue that is present in the mutated form of KRAS, but not in the normal form.[10]
History
Researchers evaluated the efficacy of sotorasib in a study of 124 participants with locally advanced or metastatic KRAS G12C-mutated non-small cell lung cancer with disease progression after receiving an immune checkpoint inhibitor and/or platinum-based chemotherapy.[2] The major outcomes measured were objective response rate (proportion of participants whose tumor is destroyed or reduced) and duration of response.[2] The objective response rate was 36% and 58% of those participants had a duration of response of six months or longer.[2]
KRAS is the most frequently mutated oncogene in cancer and encodes a key signalling protein in tumours1,2. The KRAS(G12C) mutant has a cysteine residue that has been exploited to design covalent inhibitors that have promising preclinical activity3,4,5. Here we optimized a series of inhibitors, using novel binding interactions to markedly enhance their potency and selectivity. Our efforts have led to the discovery of AMG 510, which is, to our knowledge, the first KRAS(G12C) inhibitor in clinical development. In preclinical analyses, treatment with AMG 510 led to the regression of KRASG12C tumours and improved the anti-tumour efficacy of chemotherapy and targeted agents. In immune-competent mice, treatment with AMG 510 resulted in a pro-inflammatory tumour microenvironment and produced durable cures alone as well as in combination with immune-checkpoint inhibitors. Cured mice rejected the growth of isogenic KRASG12D tumours, which suggests adaptive immunity against shared antigens. Furthermore, in clinical trials, AMG 510 demonstrated anti-tumour activity in the first dosing cohorts and represents a potentially transformative therapy for patients for whom effective treatments are lacking.
Paper
Scientific Reports (2020), 10(1), 11992
PAPER
European journal of medicinal chemistry (2021), 213, 113082.
KRAS is the most commonly altered oncogene of the RAS family, especially the G12C mutant (KRASG12C), which has been a promising drug target for many cancers. On the basis of the bicyclic pyridopyrimidinone framework of the first-in-class clinical KRASG12C inhibitor AMG510, a scaffold hopping strategy was conducted including a F–OH cyclization approach and a pyridinyl N-atom working approach leading to new tetracyclic and bicyclic analogues. Compound 26a was identified possessing binding potency of 1.87 μM against KRASG12C and cell growth inhibition of 0.79 μM in MIA PaCa-2 pancreatic cancer cells. Treatment of 26a with NCI–H358 cells resulted in down-regulation of KRAS-GTP levels and reduction of phosphorylation of downstream ERK and AKT dose-dependently. Molecular docking suggested that the fluorophenol moiety of 26a occupies a hydrophobic pocket region thus forming hydrogen bonding to Arg68. These results will be useful to guide further structural modification.
PAPER
Journal of Medicinal Chemistry (2020), 63(1), 52-65.
KRASG12C has emerged as a promising target in the treatment of solid tumors. Covalent inhibitors targeting the mutant cysteine-12 residue have been shown to disrupt signaling by this long-“undruggable” target; however clinically viable inhibitors have yet to be identified. Here, we report efforts to exploit a cryptic pocket (H95/Y96/Q99) we identified in KRASG12C to identify inhibitors suitable for clinical development. Structure-based design efforts leading to the identification of a novel quinazolinone scaffold are described, along with optimization efforts that overcame a configurational stability issue arising from restricted rotation about an axially chiral biaryl bond. Biopharmaceutical optimization of the resulting leads culminated in the identification of AMG 510, a highly potent, selective, and well-tolerated KRASG12C inhibitor currently in phase I clinical trials (NCT03600883).
The present disclosure relates to an improved, efficient, scalable process to prepare intermediate compounds, such as compound of Formula 6A, having the structure,
useful for the synthesis of compounds for the treatment of KRAS G12C mutated cancers.
BACKGROUND
[0003] KRAS gene mutations are common in pancreatic cancer, lung adenocarcinoma, colorectal cancer, gall bladder cancer, thyroid cancer, and bile duct cancer. KRAS mutations are also observed in about 25% of patients with NSCLC, and some studies have indicated that KRAS mutations are a negative prognostic factor in patients with NSCLC. Recently, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations have been found to confer resistance to epidermal growth factor receptor (EGFR) targeted therapies in colorectal cancer; accordingly, the mutational status of KRAS can provide important information prior to the prescription of TKI therapy. Taken together, there is a need for new medical treatments for patients with pancreatic cancer, lung adenocarcinoma, or colorectal cancer, especially those who have been diagnosed to have such cancers characterized by a KRAS mutation, and including those who have progressed after chemotherapy.
Related Synthetic Processes
[0126] The following intermediate compounds of 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one are representative examples of the disclosure and are not intended to be construed as limiting the scope of the present invention.
[0127] A synthesis of Compound 9 and the relevant intermediates is described in U.S. Serial No.15/984,855, filed May 21, 2018 (U.S. Publication No.2018/0334454, November 22, 2018) which claims priority to and the benefit claims the benefit of U.S. Provisional Application No.62/509,629, filed on May 22, 2017, both of which are incorporated herein by reference in their entireties for all purposes. 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one was prepared using the following process, in which the isomers of the final product were isolated via chiral chromatography.
[0128] Step 1: 2,6-Dichloro-5-fluoronicotinamide (Intermediate S). To a mixture of 2,6-dichloro-5-fluoro-nicotinic acid (4.0 g, 19.1 mmol, AstaTech Inc., Bristol, PA) in dichloromethane (48 mL) was added oxalyl chloride (2M solution in DCM, 11.9 mL, 23.8 mmol), followed by a catalytic amount of DMF (0.05 mL). The reaction was stirred at room temperature overnight and then was concentrated. The residue was dissolved in 1,4-dioxane (48 mL) and cooled to 0 °C. Ammonium hydroxide solution (28.0-30% NH3 basis, 3.6 mL, 28.6 mmol) was added slowly via syringe. The resulting mixture was stirred at 0 °C for 30 min and then was concentrated. The residue was diluted with a 1:1 mixture of EtOAc/Heptane and agitated for 5 min, then was filtered. The filtered solids were discarded, and the remaining mother liquor was partially concentrated to half volume and filtered. The filtered solids were washed with heptane and dried in a reduced-pressure oven (45 °C) overnight to provide 2,6-dichloro-5-fluoronicotinamide. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.23 (d, J = 7.9 Hz, 1 H) 8.09 (br s, 1 H) 7.93 (br s, 1 H). m/z (ESI, +ve ion): 210.9 (M+H)+.
[0129] Step 2: 2,6-Dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. To an ice-cooled slurry of 2,6-dichloro-5-fluoronicotinamide (Intermediate S, 5.0 g, 23.9 mmol) in THF (20 mL) was added oxalyl chloride (2 M solution in DCM, 14.4 mL, 28.8 mmol) slowly via syringe. The resulting mixture was heated at 75 °C for 1 h, then heating was stopped, and the reaction was concentrated to half volume. After cooling to 0 °C, THF (20 mL) was added, followed by a solution of 2-isopropyl-4-methylpyridin-3-amine (Intermediate R, 3.59 g, 23.92 mmol) in THF (10 mL), dropwise via cannula. The resulting mixture was stirred at 0 °C for 1 h and then was quenched with a 1:1 mixture of brine and saturated aqueous ammonium chloride. The mixture was extracted with EtOAc (3x) and the combined organic layers were dried over anhydrous sodium sulfate and concentrated to provide 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. This material was used without further purification in the following step. m/z (ESI, +ve ion): 385.1(M+H)+.
[0130] Step 3: 7-Chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione. To an ice-cooled solution of 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (9.2 g, 24.0 mmol) in THF (40 mL) was added KHMDS (1 M solution in THF, 50.2 mL, 50.2 mmol) slowly via syringe. The ice bath was removed and the resulting mixture was stirred for 40 min at room temperature. The reaction was quenched with saturated aqueous ammonium chloride and extracted with EtOAc (3x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% 3:1 EtOAc-EtOH/heptane) to provide 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione.1H NMR (400 MHz, DMSO-d6) δ ppm 12.27 (br s, 1H), 8.48-8.55 (m, 2 H), 7.29 (d, J = 4.8 Hz, 1 H), 2.87 (quin, J = 6.6 Hz, 1 H), 1.99-2.06 (m, 3 H), 1.09 (d, J = 6.6 Hz, 3 H), 1.01 (d, J = 6.6 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -126.90 (s, 1 F). m/z (ESI, +ve ion): 349.1 (M+H)+.
[0131] Step 4: 4,7-Dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. To a solution of 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (4.7 g, 13.5 mmol) and DIPEA (3.5 mL, 20.2 mmol) in acetonitrile (20 mL) was added phosphorus oxychloride (1.63 mL, 17.5 mmol), dropwise via syringe. The resulting mixture was heated at 80 °C for 1 h, and then was cooled to room temperature and concentrated to provide 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. This material was used without further purification in the following step. m/z (ESI, +ve ion): 367.1 (M+H)+.
[0132] Step 5: (S)-tert-Butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. To an ice-cooled solution of 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one (13.5 mmol) in acetonitrile (20 mL) was added DIPEA (7.1 mL, 40.3 mmol), followed by (S)-4-N-Boc-2-methyl piperazine (3.23 g, 16.1 mmol, Combi-Blocks, Inc., San Diego, CA, USA). The resulting mixture was warmed to room temperature and stirred for 1 h, then was diluted with cold saturated aqueous sodium bicarbonate solution (200 mL) and EtOAc (300 mL). The mixture was stirred for an additional 5 min, the layers were separated, and the aqueous layer was extracted with more EtOAc (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% EtOAc/heptane) to provide (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. m/z (ESI, +ve ion): 531.2 (M+H)+.
[0133] Step 6: (3S)-tert-Butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. A mixture of (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (4.3 g, 8.1 mmol), potassium trifluoro(2-fluoro-6-hydroxyphenyl)borate (Intermediate Q, 2.9 g, 10.5 mmol), potassium acetate (3.2 g, 32.4 mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane (661 mg, 0.81 mmol) in 1,4-dioxane (80 mL) was degassed with nitrogen for 1 min. De-oxygenated water (14 mL) was added, and the resulting mixture was heated at 90 °C for 1 h. The reaction was allowed to cool to room temperature, quenched with half-saturated aqueous sodium bicarbonate, and extracted with EtOAc (2x) and DCM (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-60% 3:1 EtOAc-EtOH/heptane) to provide (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate.1H NMR (400 MHz, DMSO-d6) δ ppm 10.19 (br s, 1 H), 8.38 (d, J = 5.0 Hz, 1 H), 8.26 (dd, J = 12.5, 9.2 Hz, 1 H), 7.23-7.28 (m, 1 H), 7.18 (d, J = 5.0 Hz, 1 H), 6.72 (d, J = 8.0 Hz, 1 H), 6.68 (t, J = 8.9 Hz, 1 H), 4.77-4.98 (m, 1 H), 4.24 (br t, J = 14.2 Hz, 1 H), 3.93-4.08 (m, 1 H), 3.84 (br d, J=12.9 Hz, 1 H), 3.52-3.75 (m, 1 H), 3.07-3.28 (m, 1 H), 2.62-2.74 (m, 1 H), 1.86-1.93 (m, 3 H), 1.43-1.48 (m, 9 H), 1.35 (dd, J = 10.8, 6.8 Hz, 3 H), 1.26-1.32 (m, 1 H), 1.07 (dd, J = 6.6, 1.7 Hz, 3 H), 0.93 (dd, J = 6.6, 2.1 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -115.65 (s, 1 F), -128.62 (s, 1 F). m/z (ESI, +ve ion): 607.3 (M+H)+.
[0134] Step 7: 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one. Trifluoroacetic acid (25 mL, 324 mmol) was added to a solution of (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (6.3 g, 10.4 mmol) in DCM (30 mL). The resulting mixture was stirred at room temperature for 1 h and then was concentrated. The residue was dissolved in DCM (30 mL), cooled to 0 °C, and sequentially treated with DIPEA (7.3 mL, 41.7 mmol) and a solution of acryloyl chloride (0.849 mL, 10.4 mmol) in DCM (3 mL; added dropwise via syringe). The reaction was stirred at 0 °C for 10 min, then was quenched with half-saturated aqueous sodium bicarbonate and extracted with DCM (2x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-100% 3:1 EtOAc-EtOH/heptane) to provide 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one.1H NMR (400 MHz, DMSO-d6) δ ppm 10.20 (s, 1 H), 8.39 (d, J = 4.8 Hz, 1 H), 8.24-8.34 (m, 1 H), 7.23-7.32 (m, 1 H), 7.19 (d, J = 5.0 Hz, 1 H), 6.87 (td, J = 16.3, 11.0 Hz, 1 H), 6.74 (d, J = 8.6 Hz, 1 H), 6.69 (t, J = 8.6 Hz, 1 H), 6.21 (br d, J = 16.2 Hz, 1 H), 5.74-5.80 (m, 1 H), 4.91 (br s, 1 H), 4.23-4.45 (m, 2 H), 3.97-4.21 (m, 1 H), 3.44-3.79 (m, 2 H), 3.11-3.31 (m, 1 H), 2.67-2.77 (m, 1 H), 1.91 (s, 3 H), 1.35 (d, J = 6.8 Hz, 3 H), 1.08 (d, J = 6.6 Hz, 3 H), 0.94 (d, J = 6.8 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ ppm -115.64 (s, 1 F), -128.63 (s, 1 F). m/z (ESI, +ve ion): 561.2 (M+H)+.
[0135] Another synthesis of Compound 9 and the relevant intermediates was described in a U.S. provisional patent application filed November 16, 2018, which is incorporated herein by reference in its entirety for all purposes.
Representative Synthetic Processes
[0136] The present disclosure comprises the following steps wherein the synthesis and utilization of the boroxine intermediate is a novel and inventive step in the manufacture of AMG 510 (Compound 9):
Raw Materials
Step la
[0137] To a solution of 2,6-dichloro-5-fluoro-3-pyridinecarboxylic acid (25kg; 119. lmol) in dichloromethane (167kg) and DMF (592g) was added Oxalyl chloride (18.9kg; 148.9mol) while maintaining an internal temp between 15-20 °C. Additional dichloromethane (33kg) was added as a rinse and the reaction mixture stirred for 2h. The reaction mixture is cooled then quenched with ammonium hydroxide (40.2L; 595.5mol) while maintaining internal temperature 0 ± 10°C. The resulting slurry was stirred for 90min then the product collected by filtration. The filtered solids were washed with DI water (3X 87L) and dried to provide 2,6-dichloro-5-fluoronicotinamide (Compound 1).
Step 1b
[0138] In reactor A, a solution of 2,6-dichloro-5-fluoronicotinamide (Compound 1) (16.27kg; 77.8mol) in dichloromethane (359.5kg) was added oxalyl chloride (11.9kg;
93.8mol) while maintaining temp ≤ 25°C for 75min. The resulting solution was then headed to 40°C ± 3°C and aged for 3h. Using vacuum, the solution was distilled to remove dichloromethane until the solution was below the agitator. Dichloromethane (300 kg) was then added and the mixture cooled to 0 ± 5°C. To a clean, dry reactor (reactor B) was added,2-isopropyl-4-methylpyridin-3-amine (ANILINE Compound 2A) (12.9kg; 85.9mol) followed by dichloromethane (102.6 kg). The ANILINE solution was azeodried via vacuum distillation while maintaining an internal temperature between 20-25 °), replacing with additional dichloromethane until the solution was dry by KF analysis (limit ≤ 0.05%). The solution volume was adjusted to approx. 23L volume with dichloromethane. The dried ANILINE solution was then added to reactor A while maintaining an internal temperature of 0 ± 5°C throughout the addition. The mixture was then heated to 23 °C and aged for 1h. the solution was polish filtered into a clean reactor to afford 2,6-dichloro-5-fluoro-N-((2- isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (Compound 3) as a solution in DCM and used directly in the next step.
Step 2
[0139] A dichloromethane solution of 2,6-dichloro-5-fluoro-N-{[4-methyl-2-(propan-2- yl)pyridin-3-yl]carbamoyl}pyridine-3-carboxamide (UREA (Compound 3)) (15kg contained; 38.9mol) was solvent exchanged into 2-MeTHF using vacuum distillation while maintaining internal temperature of 20-25 °C. The reactor volume was adjusted to 40L and then
additional 2-MeTHF was charged (105.4 kg). Sodium t-butoxide was added (9.4 kg;
97.8mol) while maintaining 5-10 °C. The contents where warmed to 23 °C and stirred for 3h. The contents where then cooled to 0-5C and ammonium chloride added (23.0kg; 430mol) as a solution in 60L of DI water. The mixture was warmed to 20 C and DI water added (15L) and further aged for 30min. Agitation was stopped and the layers separated. The aqueous layer was removed and to the organic layer was added DI water(81.7L). A mixture of conc HCl (1.5kg) and water (9L) was prepared then added to the reactor slowly until pH measured between 4-5. The layers were separated, and the aqueous layer back extracted using 2-MeTHF (42.2kg). The two organic layers combined and washed with a 10% citric acid solution (75kg) followed by a mixture of water (81.7L) and saturated NaCl (19.8 kg). The organic layer was then washed with saturated sodium bicarbonate (75kg) repeating if necessary to achieve a target pH of ≥ 7.0 of the aqueous. The organic layer was washed again with brine (54.7kg) and then dried over magnesium sulfate (5kg). The mixture was filtered to remove magnesium sulfate rinsing the filtered bed with 2-MeTHF (49.2 kg). The combined filtrate and washes where distilled using vacuum to 40L volume. The concentrated solution was heated to 55 °C and heptane (10-12kg) slowly added until cloud point. The solution was cooled to 23 °C over 2h then heptane (27.3 kg) was added over 2h. The product slurry was aged for 3h at 20-25 °C then filtered and washed with a mixture of 2-MeTHF (2.8kg) and heptane (9kg). The product was dried using nitrogen and vacuum to afford solid 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (rac-DIONE (Compound 4)).
Step 3
[0140] To a vessel, an agitated suspension of Compound 4, (1.0 eq.) in 2- methylterahydrofuran (7.0 L/kg) was added (+)-2,3-dibenzoyl-D-tartaric acid (2.0 eq.) under an atmosphere of nitrogen. 2-MeTHF is chiral, but it is used as a racemic mixture. The different enantiomers of 2-MeTHF are incorporated randomly into the co-crystal. The resulting suspension was warmed to 75°C and aged at 75°C until full dissolution was observed (< 30 mins.). The resulting solution was polish filtered at 75°C into a secondary vessel. To the polish filtered solution was charged n-Heptane (2.0 L/kg) at a rate that maintained the internal temperature above 65°C. The solution was then cooled to 60°C, seeded with crystals (0.01 kg/kg) and allowed to age for 30 minutes. The resulting suspension was cooled to 20°C over 4 hours and then sampled for chiral purity analysis by HPLC. To the suspension, n-Heptane (3.0 L/kg) was charged and then aged for 4 hours at 20°C under an atmosphere of nitrogen. The suspension was filtered, and the isolated solids were washed two times with (2:1) n-Heptane:2-methyltetrahydrofuran (3.0 L/kg). The material was dried with nitrogen and vacuum to afford M-Dione:DBTA: Me-THF complex (Compound 4a).
Step 4
[0141] To vessel A, a suspension of disodium hydrogen phosphate (21.1 kg, 2.0 equiv) in DI water (296.8 L, 6.3 L/kg) was agitated until dissolution was observed (≥ 30 min.). To vessel B, a suspension of the M-Dione:DBTA: Me-THF complex (Composition 4a)[46.9 kg (25.9 kg corrected for M-dione, 1.0 equiv.)] in methyl tert-butyl ether (517.8 L, 11.0 L/kg) was agitated for 15 to 30 minutes. The resulting solution from vessel A was added to vessel B, and then the mixture was agitated for more than 3 hours. The agitation was stopped, and the biphasic mixture was left to separate for more than 30 minutes. The lower aqueous phase was removed and then back extracted with methyl tert-butyl ether (77.7 L, 1.7 L/kg). The organic phases were combined in vessel B and dried with magnesium sulfate (24.8 kg, 0.529 kg/kg). The resulting suspension from vessel B was agitated for more than three hours and then filtered into vessel C. To vessel B, a methyl tert-butyl ether (46.9 L, 1.0 L/kg) rinse was charged and then filtered into vessel C. The contents of vessel C were cooled to 10 °C and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until 320-350 kg (6.8-7.5 kg/kg) of methyl tert-butyl ether was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged a second time over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (195.9 L, 4.2 L/kg) was charged a third time over one hour and then sampled for solvent composition by GC analysis. The vessel C suspension continued to agitate for more than one hour. The suspension was filtered, and then washed with a n-Heptane (68.6 L, 1.5 L/kg) rinse from vessel C. The isolated solids were dried at 50°C, and a sample was submitted for stock suitability. Afforded 7-chloro-6-fluoro-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (M-DIONE) Compound 5M.
[0142] The first-generation process highlighted above has been successfully scaled on 200+ kg of rac-dione starting material (Compound 4). In this process, seeding the crystallization with the thermodynamically-stable rac-dione crystal form (which exhibits low solubility) would cause a batch failure. Based on our subsequent studies, we found that increasing the DBTA equivalents and lowering the seed temperature by adjusting heptane
charge schedule improves robustness of the process. The improved process is resistant to the presence of the thermodynamically-stable rac-dione crystal form and promotes successful separation of atropisomers. Subsequent batches will incorporate the improved process for large scale manufacture.
Step 5
Note: All L/kg amounts are relative to M-Dione input; All equiv. amounts are relative to M-Dione input after adjusted by potency.
[0143] M-Dione (Compound 5M, 1.0 equiv.) and Toluene-1 (10.0 L/kg) was charged to Vessel A. The resulting solution was dried by azeotropic distillation under vacuum at 45 °C until 5.0 L/kg of solvents has been removed. The contents of Vessel A were then cooled to 20 °C.
[0144] Vessel C was charged with Toluene-3 (4.5 L/kg), Phosphoryl chloride (1.5 equiv.) and N,N-Diisopropylethylamine-1 (2.0 equiv.) while maintaining the internal temperature below 20 ± 5 °C.
Upon finishing charging, Vessel C was warmed to 30 ± 5 °C. The contents of Vessel A were then transferred to Vessel C over 4 hours while maintaining the internal temperature at 30 ± 5°C. Vessel A was rinsed with Toluene-2 (0.5 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated at 30°C for an additional 3 hours. The contents of Vessel C were cooled to 20 ± 5 °C. A solution of (s)-1-boc-3-methylpiperazine (1.2 equiv.), N,N-Diisopropylethylamine-2 (1.2 equiv.) in isopropyl acetate-1 (1.0 L/kg) was prepared in Vessel D. The solution of Vessel D was charged to vessel C while maintaining a batch temperature of 20 ± 5 °C (Note: Exotherm is observed). Upon the end of transfer, Vessel D was rinsed with additional dichloromethane (1.0 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated for an additional 60 minutes at 20 °C. A solution of sodium bicarbonate [water-1 (15.0 L/kg + Sodium bicarbonate (4.5 equiv.)] was then charged into Vessel C over an hour while maintaining an internal temperature at 20 ± 5 °C throughout the addition. The contents of Vessel C were agitated for at least 12 hours at which point the Pipazoline (Compound 6) product was isolated by filtration in an agitated filter dryer. The cake was washed with water-2 and -3 (5.0 L/kg x 2 times, agitating each wash for 15 minutes) and isopropyl acetate-2 and 3 (5.0 L/kg x 2 times, agitating each wash for 15 min). The cake as dried under nitrogen for 12 hours.
Acetone Re-slurry (Optional):
[0145] Pipazoline (Compound 6) and acetone (10.0 L/kg) were charged to Vessel E. The suspension was heated to 50 °C for 2 hours. Water-4 (10.0 L/kg) was charged into Vessel E over 1 hour. Upon completion of water addition, the mixture was cooled to 20 °C over 1 hour. The contents of Vessel E were filtered to isolate the product, washing the cake with 1:1 acetone/water mixture (5.0 L/kg). The cake was dried under nitrogen for 12 hours.
Step 6
General Note: All equivalents and volumes are reported in reference to Pipazoline input
Note: All L/kg and kg/kg amounts are relative to Pipazoline input
[0146] Reactor A is charged with Pipazoline (Compound 6, 1.0 equiv), degassed 2- MeTHF (9.0 L/kg) and a solution of potassium acetate (2.0 equiv) in degassed water (6.5 L/kg). The resulting mixture is warmed to 75 ± 5 °C and then, charge a slurry of
Pd(dpePhos)Cl2 (0.003 equiv) in 2-MeTHF (0.5 L/kg). Within 2 h of catalyst charge, a solution of freshly prepared Boroxine (Compound 6A, 0.5 equiv) in wet degassed 2-MeTHF (4.0 L/kg, KF > 4.0%) is charged over the course of >1 hour, but < 2 hours, rinsing with an additional portion of wet 2-MeTHF (0.5 L/kg) after addition is complete. After reaction completion ( <0.15 area % Pipazoline remaining, typically <1 h after boroxine addition is complete), 0.2 wt% (0.002 kg/kg) of Biaryl seed is added as a slurry in 0.02 L/kg wet 2- MeTHF, and the resulting seed bed is aged for > 60 min. Heptane (5.0 L/kg) is added over 2 hours at 75 ± 5 °C. The batch is then cooled to 20 ± 5 °C over 2 hours and aged for an additional 2 h. The slurry is then filtered and cake washed with 1 x 5.0L/kg water, 1 x 5.0L/kg 1:1 iPrOH:water followed by 1 x 5.0 L/kg 1:1 iPrOH:heptane (resuspension wash: the cake is resuspended by agitator and allow to set before filtering) . The cake (Biaryl, Compound 7) is then dried under vacuum with a nitrogen sweep.
Note: If the reaction stalls, an additional charge of catalyst and boroxine is required
Step 7 Charcoal Filtration for Pd removal
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to crude Biaryl input
[0147] In a clean Vessel A, charge crude Biaryl (1 equiv) and charge DCM (10 L/kg). Agitate content for > 60 minutes at 22 ± 5 °C, observing dissolution. Pass crude Biaryl from Vessel A, through a bag filter and carbon filters at a flux ≤ 3 L2/min/m and collect filtrate in clean Vessel B. Charge DCM rinse (1 L/kg) to Vessel A, and through carbon filters to collect in vessel B.
[0148] From filtrate in Vessel B, pull a solution sample for IPC Pd content. Sample is concentrated to solid and analyzed by ICP-MS. IPC: Pd ≤ 25 ppm with respect to Biaryl. a. If Pd content is greater than 25 ppm with respect to Biaryl on first or second IPC sample, pass solution through carbon filter a second time at ≤ 3 L2/min/m2, rinsing with 1 L/kg DCM; sample filtrate for IPC.
b. If Pd content remains greater than 25 ppm after third IPC, install and condition fresh carbon discs. Pass Biaryl filtrate through refreshed carbon filter, washing with 1 L/kg DCM. Sample for IPC.
[0149] Distill and refill to appropriate concentration. Prepare for distillation of recovered filtrate by concentrating to ≤ 4 L/kg DCM, and recharge to reach 5.25 ± 0.25 L/kg DCM prior to moving into Step 7 Boc-deprotection reaction.
Step 7
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to Biaryl input
[0150] To Reactor A was added: tert-butyl (3S)-4-{6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl}-3-methylpiperazine-1-carboxylate (Biaryl) (1.0 equiv), dichloromethane (5.0 L/kg), and the TFA (15.0 equiv, 1.9 L/kg) is charged slowly to maintain the internal temperature at 20 ± 5 °C. The reaction was stirred for 4 h at 20 ± 5 °C.
[0151] To Reactor B was added: potassium carbonate (18.0 equiv), water (20.0 L/kg), and NMP (1.0) to form a homogenous solution. While agitating at the maximum acceptable rate for the equipment, the reaction mixture in A was transferred into the potassium carbonate solution in B over 30 minutes (~ 0.24 L/kg/min rate). The mixture was stirred at 20 ± 5 °C for an additional 12 h.
[0152] The resulting slurry was filtered and rinsed with water (2 x 10 L/kg). The wet cake was dried for 24 h to give 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-4-[(2S)-2-methylpiperazin- 1-yl]-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Des- Boc, Compound 8).
Step 8
Note: All L/kg and kg/kg amounts are relative to Des-Boc input
[0153] Des-Boc (Compound 8, 1.0 equiv) and NMP (4.2 L/kg) are charged to Vessel A under nitrogen, charge the TFA (1.0 equiv.) slowly to maintain the Tr <25 °C. The mixture is aged at 25 °C until full dissolution is observed (about 0.5 hour). The solution is then polish filtered through a 0.45 micron filter into Vessel B, washing with a NMP (0.8 L/kg). The filtrate and wash are combined, and then cooled to 0 °C. To the resulting solution, Acryloyl Chloride (1.3 equiv.) is added while maintaining temperature < 10 C. The reaction mixture is then aged at 5 ±5°C until completed by IPC (ca.1.5 hrs).
Preparation of Aqueous Disodium Phosphate Quench:
[0154] Disodium Phosphate (3.0 equiv) and Water (15.0 L/kg) are charged to Vessel C. The mixture is aged at 25 °C until full dissolution is observed. The solution is warmed to 45 ±5°C. A seed slurry of AMG 510 (0.005 equiv.) in Water (0.4 L/kg) is prepared and added to Vessel C while maintaining temperature at 45 ±5°C.
[0155] The reaction mixture in Vessel B is transferred to Vessel C (quench solution) while maintaining temperature at 45 ±5°C (ca.1 hrs). Vessel B is washed with a portion of NMP (0.5 L/kg). The product slurry is aged for 2 hrs at 45 ±5°C, cooled to 20 °C over 3 hrs, aged at 20 °C for a minimum of 12 hrs, filtered and washed with Water (2 x 10.0 L/kg). The product is dried using nitrogen and vacuum to afford Crude AMG 510 (Compound 9A).
Step 9
General Note: All equivalents and volumes are reported in reference to crude AMG 510 input
Note: All L/kg and kg/kg amounts are relative to Crude AMG 510 input
[0156] Reactor A was charged with 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4- methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1- yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Crude AMG 510) (1.0 equiv), ethanol (7.5 L/kg), and water (1.9 L/kg). The mixture heated to 75 °C and polish filtered into a clean Reactor B. The solution was cool to 45 °C and seeded with authentic milled AMG 510 seed (0.015 േ 0.005
1 Seed performs best when reduced in particle size via milling or with other type of mechanical grinding if mill is not available (mortar/ pestle). Actual seed utilized will be based on seed availability. 1.0- 2.0% is seed is target amount.
kg/kg); the resulting slurry was aged for 30 min. Water (15.0 L/kg) was added over 5h while maintaining an internal temperature > 40 °C; the mixture was aged for an additional 2h.
[0157] The mixture was cooled to 20 °C over 3 hours and aged for 8h, after which the solid was collected by filtration and washed using a mixture of ethanol (2.5 L/kg) and water (5.0 L/kg). The solid was dried using vacuum and nitrogen to obtain 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510, Compound 9).
Compound 6A Boroxine Synthesis:
Lithiation/borylation
[0158] Reactor A was charged with THF (6 vol), a secondary amine base, Diisopropylamine (1.4 equiv), and a catalyst, such as triethylamine hydrochloride (0.01 equiv.). The resulting solution was cooled to -70 °C and a first base, n-BuLi (2.5 M in hexane, 1.5 equiv) was slowly added. After addition is complete, a solution of 3-fluoroanisole (1.0 equiv) in THF (6 vol) was added slowly and kept at -70 °C for 5 min. Concurrently or subsequently, a reagent, B(EtO)3 (2.0 equiv), was added slowly and kept at -70 °C for 10 min. The reaction mixture was quenched with an acid, 2N HCl. The quenched reaction mixture was extracted with MTBE (3 x 4 vol). The combined organic phases were concentrated to 1.5-3 total volumes. Heptane (7-9 vol) was added drop-wise and the mixture was cooled to 0-10 °C and stirred for 3 h. The mixture was filtrated and rinsed with heptane (1.5 vol). The solid was dried under nitrogen at < 30 °C to afford (2-fluoro-6-methoxyphenyl)boronic acid.
Demethylation:
Note: All L/kg and kg/kg amounts are relative to (2-fluoro-6-methoxyphenyl)boronic acid input
[0159] To a reactor, charge dichloromethane (solvent, 4.0 L/kg) and an acid, BBr3 (1.2 equiv), and cool to -20 °C. To this solution, a suspension of (2-fluoro-6-methoxyphenyl)boronic acid (1.0 equiv) in dichloromethane (4.0 L/kg) was added into the BBr3/DCM mixture while keeping temperature -15 to -25 °C. The reaction was allowed to proceed for approximately 2 hours while monitored by HPLC [≤1% (2-fluoro-6-methoxyphenyl)boronic acid] before reverse quenching into water (3.0 L/kg). The precipitated solid was then isolated by filtration and slurried with water (3.0 L/kg) on the filter prior to deliquoring. The filtrates were adjusted to pH 4-6 by the addition of sodium bicarbonate. The bottom organic phase was separated and the resulting aqueous layer was washed with dichloromethane (solvent, 5.0 Vol) and adjusted to pH = 1 by addition of concentrated hydrochloric acid. The resulting solids were isolated by filtration, washing the cake with water (2 x 5.0 L/kg)
Purification via Reslurry (required)
[0160] The combined crude solids were charged into a reactor and slurried with 5% EtOH/water (5.0 L/kg) at 20 °C for >1 h. The purified product was then isolated by filtration and rinsed with water (2 x 3 L/kg) before drying on the filter at < 30 °C to with nitrogen/vacuum to afford 2,2′,2”-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tris(3-fluorophenol) (Boroxine, Compound 6A).
^ Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation ” at ClinicalTrials.gov
“Sotorasib”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation (CodeBreaK 100)” at ClinicalTrials.gov
PLAIN F 1423758-00-2 WITHOUT RADIO LABELC18 H23 F N4 O8, 441.4L-Glutamic acid, N-[[[(1S)-1-carboxy-5-[[[6-(fluoro-18F)-3-pyridinyl]carbonyl]amino]pentyl]amino]carbonyl]-2-(3-{1-carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl) amino]-pentyl}ureido)-pentanedioic acid
For positron emission tomography imaging of prostate-specific membrane antigen-positive lesions in men with prostate cancer
For positron emission tomography (PET) of prostatespecific membrane antigen (PSMA) positive lesions in men with prostate cancer: • with suspected metastasis who are candidates for initial definitive therapy. • with suspected recurrence based on elevated serum prostate-specific antigen (PSA) level.
Originator Johns Hopkins University School of Medicine
Class Amides; Carboxylic acids; Fluorinated hydrocarbons; Imaging agents; Pyridines; Radiopharmaceutical diagnostics; Radiopharmaceuticals; Small molecules; Urea compounds
Mechanism of ActionPositron-emission tomography enhancers
Orphan Drug StatusNo
MarketedProstate cancer
28 May 2021Registered for Prostate cancer (Diagnosis) in USA (IV) – First global approval
28 May 2021Adverse events data from phase III CONDOR and phase II/III OSPREY trials in prostate cancer released by Lantheus Holdings
27 May 2021Lantheus Holdings intends to launch Fluorine-18 DCFPyL in USA at end of 2021
PYLARIFY contains fluorine 18 (F 18), radiolabeled prostate-specific membrane antigen inhibitor imaging agent. Chemically piflufolastat F 18 is 2-(3-{1-carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl) amino]-pentyl}ureido)-pentanedioic acid. The molecular weight is 441.4 and the structural formula is:
The chiral purity of the unlabeled piflufolastat F 18 precursor is greater than 99% (S,S). PYLARIFY is a sterile, non-pyrogenic, clear, colorless solution for intravenous injection. Each milliliter contains 37 to 2,960 MBq (1 to 80 mCi) piflufolastat F 18 with ≤0.01 µg/mCi of piflufolastat at calibration time and date, and ≤ 78.9 mg ethanol in 0.9% sodium chloride injection USP. The pH of the solution is 4.5 to 7.0. PYLARIFY has a radiochemical purity of at least 95% up to 10 hours following end of synthesis, and specific activity of at least 1000 mCi/µmol at the time of administration.
PYLARIFY contains fluorine 18 (F 18), radiolabeled prostate-specific membrane antigen inhibitor imaging agent. Chemically piflufolastat F 18 is 2-(3-{1-carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl)amino]-pentyl}ureido)-pentanedioic acid. The molecular weight is 441.4 and the structural formula is:
The chiral purity of the unlabeled piflufolastat F 18 precursor is greater than 99% (S,S).
PYLARIFY is a sterile, non-pyrogenic, clear, colorless solution for intravenous injection. Each milliliter contains 37 to 2,960 MBq (1 to 80 mCi) piflufolastat F 18 with ≤0.01 μg/mCi of piflufolastat at calibration time and date, and ≤ 78.9 mg ethanol in 0.9% sodium chloride injection USP. The pH of the solution is 4.5 to 7.0.
PYLARIFY has a radiochemical purity of at least 95% up to 10 hours following end of synthesis, and specific activity of at least 1000 mCi/μmol at the time of administration.
Physical Characteristics
PYLARIFY is radiolabeled with fluorine 18 (F 18), a cyclotron produced radionuclide that decays by positron emission to stable oxygen 18 with a half-life of 109.8 minutes. The principal photons useful for diagnostic imaging are the coincident pair of 511 keV gamma photons, resulting from the interaction of the emitted positron with an electron (Table 3).
Table 3: Principal Radiation Produced from Decay of Fluorine 18
US FDA APPROVED 9/15/2021, Exkivity, To treat locally advanced or metastatic non-small cell lung cancer with epidermal growth factor receptor exon 20 insertion mutation
On September 15, 2021, the Food and Drug Administration granted accelerated approval to mobocertinib (Exkivity, Takeda Pharmaceuticals, Inc.) for adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.
Today, the FDA also approved the Oncomine Dx Target Test (Life Technologies Corporation) as a companion diagnostic device to select patients with the above mutations for mobocertinib treatment.
Approval was based on Study 101, an international, non-randomized, open-label, multicohort clinical trial (NCT02716116) which included patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations. Efficacy was evaluated in 114 patients whose disease had progressed on or after platinum-based chemotherapy. Patients received mobocertinib 160 mg orally daily until disease progression or intolerable toxicity.
The main efficacy outcome measures were overall response rate (ORR) according to RECIST 1.1 as evaluated by blinded independent central review (BICR) and response duration. The ORR was 28% (95% CI: 20%, 37%) with a median response duration of 17.5 months (95% CI: 7.4, 20.3).
The most common adverse reactions (>20%) were diarrhea, rash, nausea, stomatitis, vomiting, decreased appetite, paronychia, fatigue, dry skin, and musculoskeletal pain. Product labeling includes a boxed warning for QTc prolongation and Torsades de Pointes, and warnings for interstitial lung disease/pneumonitis, cardiac toxicity, and diarrhea.
The recommended mobocertinib dose is 160 mg orally once daily until disease progression or unacceptable toxicity.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), and United Kingdom’s Medicines & Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 6 weeks ahead of the FDA goal date.
Approval based on Phase 1/2 trial results, which demonstrated clinically meaningful responses with a median duration of response (DoR) of approximately 1.5 years
Next-generation sequencing (NGS) companion diagnostic test approved simultaneously to support identification of patients with EGFR Exon20 insertion mutations
OSAKA, Japan, and CAMBRIDGE, Mass. September 15, 2021 – Takeda Pharmaceutical Company Limited (TSE:4502/NYSE:TAK) (“Takeda”) today announced that the U.S. Food and Drug Administration (FDA) has approved EXKIVITY (mobocertinib) for the treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy. EXKIVITY, which was granted priority review and received Breakthrough Therapy Designation, Fast Track Designation and Orphan Drug Designation from the FDA, is the first and only approved oral therapy specifically designed to target EGFR Exon20 insertion mutations. This indication is approved under Accelerated Approval based on overall response rate (ORR) and DoR. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
“The approval of EXKIVITY introduces a new and effective treatment option for patients with EGFR Exon20 insertion+ NSCLC, fulfilling an urgent need for this difficult-to-treat cancer,” said Teresa Bitetti, president, Global Oncology Business Unit, Takeda. “EXKIVITY is the first and only oral therapy specifically designed to target EGFR Exon20 insertions, and we are particularly encouraged by the duration of the responses observed with a median of approximately 1.5 years. This approval milestone reinforces our commitment to meeting the needs of underserved patient populations within the oncology community.”
The FDA simultaneously approved Thermo Fisher Scientific’s Oncomine Dx Target Test as an NGS companion diagnostic for EXKIVITY to identify NSCLC patients with EGFR Exon20 insertions. NGS testing is critical for these patients, as it can enable more accurate diagnoses compared to polymerase chain reaction (PCR) testing, which detects less than 50% of EGFR Exon20 insertions.
“EGFR Exon20 insertion+ NSCLC is an underserved cancer that we have been unable to target effectively with traditional EGFR TKIs,” said Pasi A. Jänne, MD, PhD, Dana Farber Cancer Institute. “The approval of EXKIVITY (mobocertinib) marks another important step forward that provides physicians and their patients with a new targeted oral therapy specifically designed for this patient population that has shown clinically meaningful and sustained responses.”
“Patients with EGFR Exon20 insertion+ NSCLC have historically faced a unique set of challenges living with a very rare lung cancer that is not only underdiagnosed, but also lacking targeted treatment options that can improve response rates,” said Marcia Horn, executive director, Exon 20 Group at ICAN, International Cancer Advocacy Network. “As a patient advocate working with EGFR Exon20 insertion+ NSCLC patients and their families every day for nearly five years, I am thrilled to witness continued progress in the fight against this devastating disease and am grateful for the patients, families, healthcare professionals and scientists across the globe who contributed to the approval of this promising targeted therapy.”
The FDA approval is based on results from the platinum-pretreated population in the Phase 1/2 trial of EXKIVITY, which consisted of 114 patients with EGFR Exon20 insertion+ NSCLC who received prior platinum-based therapy and were treated at the 160 mg dose. Results were presented at the 2021 American Society of Clinical Oncology (ASCO) Annual Meeting from the Phase 1/2 trial and demonstrated a confirmed ORR of 28% per independent review committee (IRC) (35% per investigator) as well as a median DoR of 17.5 months per IRC, a median overall survival (OS) of 24 months and a median progression-free survival (PFS) of 7.3 months per IRC.
The most common adverse reactions (>20%) were diarrhea, rash, nausea, stomatitis, vomiting, decreased appetite, paronychia, fatigue, dry skin, and musculoskeletal pain. The EXKIVITY Prescribing Information includes a boxed warning for QTc prolongation and Torsades de Pointes, and warnings and precautions for interstitial lung disease/pneumonitis, cardiac toxicity, and diarrhea.
The FDA review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence (OCE), which provides a framework for concurrent submission and review of oncology products among international partners. We look forward to continuing our work with regulatory agencies across the globe to bring mobocertinib to patients.
About EXKIVITY (mobocertinib)
EXKIVITY is a first-in-class, oral tyrosine kinase inhibitor (TKI) specifically designed to selectively target epidermal growth factor receptor (EGFR) Exon20 insertion mutations.
EXKIVITY is approved in the U.S. for the treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with EGFR exon 20 insertion mutations as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.
Results from the Phase 1/2 trial of mobocertinib have also been accepted for review by the Center for Drug Evaluation (CDE) in China for locally advanced or metastatic NSCLC patients with EGFR Exon20 insertion mutations who have been previously treated with at least one prior systemic chemotherapy.
Non-small cell lung cancer (NSCLC) is the most common form of lung cancer, accounting for approximately 85% of the estimated 2.2 million new cases of lung cancer diagnosed each year worldwide, according to the World Health Organization.1,2 Patients with epidermal growth factor receptor (EGFR) Exon20 insertion+ NSCLC make up approximately 1-2% of patients with NSCLC, and the disease is more common in Asian populations compared to Western populations.3-7 This disease carries a worse prognosis than other EGFR mutations, as EGFR TKIs – which do not specifically target EGFR Exon20 insertions – and chemotherapy provide limited benefit for these patients.
Takeda is committed to continuing research and development to meet the needs of the lung cancer community through the discovery and delivery of transformative medicines.
EXKIVITY IMPORTANT SAFETY INFORMATION
QTc Interval Prolongation and Torsades de Pointes: EXKIVITY can cause life-threatening heart rate-corrected QT (QTc) prolongation, including Torsades de Pointes, which can be fatal, and requires monitoring of QTc and electrolytes at baseline and periodically during treatment. Increase monitoring frequency in patients with risk factors for QTc prolongation. Avoid use of concomitant drugs which are known to prolong the QTc interval and use of strong or moderate CYP3A inhibitors with EXKIVITY, which may further prolong the QTc. Withhold, reduce the dose, or permanently discontinue EXKIVITY based on the severity of QTc prolongation.
Interstitial Lung Disease (ILD)/Pneumonitis: Monitor patients for new or worsening pulmonary symptoms indicative of ILD/pneumonitis. Immediately withhold EXKIVITY in patients with suspected ILD/pneumonitis and permanently discontinue EXKIVITY if ILD/pneumonitis is confirmed.
Cardiac Toxicity: Monitor cardiac function, including left ventricular ejection fraction, at baseline and during treatment. Withhold, resume at reduced dose or permanently discontinue based on severity.
Diarrhea: Diarrhea may lead to dehydration or electrolyte imbalance, with or without renal impairment. Monitor electrolytes and advise patients to start an antidiarrheal agent at first episode of diarrhea and to increase fluid and electrolyte intake. Withhold, reduce the dose, or permanently discontinue EXKIVITY based on the severity.
Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective non-hormonal contraception.
The most common side effects include diarrhea, rash, nausea, stomatitis, vomiting, decreased appetite, paronychia, fatigue, dry skin, and musculoskeletal pain.[2]
Mobocertinib was approved for medical use in the United States in September 2021.[2][3] It is a first-in-class oral treatment to target EGFR Exon20 insertion mutations.[3]
Medical uses
Mobocertinib is indicated for adults with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.[2]
Example 1 Procedure for the preparation of isopropyl 2-((5-acrylamido-4-((2- (dimethylamino)ethyl) (methyl)amino)-2-methoxyphenyl)amino)-4-(l -methyl- lH-indol-3- yl)pyrimidine-5-carboxylate (Compound (A)).
[00352] To a 2 L Radley reactor equipped with a mechanical stirrer, a thermometer, and a refluxing condenser was charged isopropyl 2,4-dichloropyrimidine-5-carboxylate (100 g, 42.5 mmol, 1.00 eq.) andl,2-dimethoxyethane (DME, 1.2 L, 12 vol) at RT. The mixture was cooled to 3 °C, and granular AlCb (65.5 g, 49.1 mmol, 1.15 eq.) was added in 2 portions (IT 3-12 °C, jacket set 0 °C). The white slurry was stirred 15-25 °C for 60 minutes. 1 -Methylindole (59 g, 44.9 mmol, 1.06 eq.) was added in one portion (IT 20-21°C). DME (100 mL) was used to aid 1- Methylindole transfer. The reaction mixture was aged for at 35 °C for 24 h. Samples (1 mL) were removed at 5 h and 24 h for HPLC analysis (TM1195).[00353] At 5 h the reaction had 70 % conversion, while after 24 h the desired conversion was attained (< 98%).[00354] The reaction mixture was cooled to 0 °C to 5 °C and stirred for 1 h. The solids were collected via filtration and washed with DME (100 mL). The solids (AlCb complex) were charged back to reactor followed by 2-MeTHF (1 L, 10 vol), and water (400 mL, 4 vol). The mixture was stirred for 10 minutes. The stirring was stopped to allow the layers to separate.The organic phase was washed with water (200 mL, 2 vol). The combined aqueous phase was re-extracted with 2-MeTHF (100 mL, 1 vol).[00355] During workup a small amount of product title compound started to crystallize.Temperature during workup should be at about 25-40 °C.[00356] The combined organic phase was concentrated under mild vacuum to 300-350 mL (IT 40-61 °C). Heptane (550 mL) was charged while maintaining the internal temperature between 50 °C and 60 °C. The resulting slurry was cooled at 25 °C over 15 minutes, aged for 1 h (19-25 °C) and the resulting solids isolated by filtration.[00357] The product was dried at 50 °C under vacuum for 3 days to yield 108.1 g (77 % yield) of the title compound, in 100% purity (AUC) as a yellow solid.‘H NMR (400 MHz, DMSO-i/e) d ppm 1.24 (d, J= 6.53 Hz, 6 H) 3.92 (s, 3 H) 5.19 (spt, J=6.27 Hz, 1 H) 7.25 – 7.35 (m, 2 H) 7.59 (d, J=8.03 Hz, 1 H) 8.07 (s, 1 H) 8.16 (d, J= 7.53 Hz, 1 H) 8.82 (s, 1 H).[00358] Step 2: Preparation of isopropyl 2-((4-fhioro-2-methoxy-5-nitrophenyl)amino)-4-(l- methyl-lH-indol-3-yl)pyrimidine-5-carboxylate.
[00359] A mixture of the product of step 1 (85.0 g, 258 mmol, 1.0 eq.), 4-fluoro-2-methoxy- 5nitroaniline (57.0 g, 306 mmol, 1.2 eq.) and PTSA monohydrate (13.3 g, 70.0 mmol, 0.27 eq.) in acetonitrile (1.4 L, 16.5 v) was heated to 76-81 °C under nitrogen in a 2 L Radley reactor. IPC at 19 h indicated that the reaction was complete.[00360] The reaction mixture was cooled to 25 °C and water (80 mL) was charged in one portion (IT during charge dropped from 25 °C to 19 °C). The reaction mixture was aged for 1 h at 21 °C and then the resulting solids were isolated by filtration. The product was washed with EtOAc (2 x 150 mL) and dried in high vacuum at 50 °C to 60 °C for 44 h to give 121.5 g of the title compound (98% yield), HPLC purity 100 % a/a; NMR indicated that PTSA was purged.¾ NMR (400 MHz, DMSO-7,) d ppm 1.21 (d, 7=6.02 Hz, 6 H) 3.91 (s, 3 H) 4.02 (s, 3 H) 5.09 (spt, 7=6.27 Hz, 1 H) 7.10 (t, 7=7.53 Hz, 1 H) 7.26 (t, 7=7.58 Hz, 1 H) 7.42 (d, 7=13.05 Hz, 1 H) 7.55 (d, 7=8.53 Hz, 1 H) 7.90 (br d, 7=7.53 Hz, 1 H) 7.98 (s, 1 H) 8.75 (s, 1 H) 8.88 (d, 7=8.03 Hz, 1 H) 9.03 (s, 1 H).[00361] Step 3: Preparation of isopropyl 2-((4-((2-(dimethylamino)ethyl(methyl)amino)-2- methoxy-5-nitrophenyl)amino)-4-(l-methyl-lH-indol-3-yl)pyrimidine-5-carboxylate.
[00362] A 50 L flask was charged 1.500 kg of the product of step 2 (3.1 moles, l.O equiv.), 639.0 g A,A,A-trimethylethylenediamine (6.3 mol, 2 equiv.), and 21 L MeCN. The resulting slurry was mixed for 7 hours at reflux. The reaction was cooled overnight. Water (16.5 L) was added before the solids were isolated. After isolation of the solids, a wash of 2.25 L MeCN in 2.25 L water was conducted to provide the title compound. The solids were dried, under vacuum, at 75 °C. HPLC purity a/a % of the dry solid was 99.3%.¾ NMR (400 MHz, DMSO-7,) d ppm 1.22 (d, 7=6.02 Hz, 6 H) 2.09 – 2.13 (m, 1 H) 2.19 (s, 6 H) 2.49 – 2.52 (m, 1 H) 2.89 (s, 3 H) 3.29 – 3.35 (m, 2 H) 3.89 (s, 3 H) 3.94 (s, 3 H) 5.10 (spt, 7=6.19 Hz, 1 H) 6.86 (s, 1 H) 7.07 (br t, 7=7.53 Hz, 1 H) 7.24 (t, 7=7.28 Hz, 1 H) 7.53 (d, 7=8.53Hz, 1 H) 7.86 – 8.02 (m, 2 H) 8.36 (s, 1 H) 8.69 (s, 1 H) 8.85 (s, 1 H).[00363] Step 4: Preparation of isopropyl 2-((5-amino-4-((2-(dimethylamino)ethyl)(methyl)- amino)-2-methoxyphenyl)amino)-4-(l -methyl- lH-indo 1-3 -yl)pyrimidine-5-carboxy late.
[00364] To a mixture of the product of step 3 (1.501 kg, 2.67 mol, 1.00 eq.) and 10% Pd/C (64 % wet, 125.0 g, 0.01 1 eq.) was added 2-MeTHF (17.7 L) in a 20 L pressure reactor. The mixture was hydrogenated at 6- 10 psi ¾ and at 40 °C until IPC indicated complete conversion (1 1 h, the reaction product 99.0%). The reaction mixture was filtered (Celite), and the pad rinsed with MeTHF (2.5 L total). The filtrate was stored under N2 in a refrigerator until crystallization.[00365] Approximately 74% of 2-MeTHF was evaporated under reduced pressure while maintaining IT 23-34 °C (residual volume in the reactor was approximately 4.8 L). To the mixture was added n-heptane (6 L) over 15 min via dropping funnel. The resulting slurry was aged at room temperature overnight. The next day the solids on the walls were scraped to incorporate them into the slurry and the solids were isolated by filtration. The isolated solids were washed with n-heptane containing 5% MeTFlF (2 x 750 mL), and dried (75 °C, 30 inch Flg) to yield 1287 g (91 % yield) of the title compound as a yellow solid. F1PLC purity: 99.7% pure.[00366] ¾ NMR (400 MHz, DMSO- ) d ppm 1.20 (d, .7=6.02 Hz, 6 H) 2.21 (s, 6 H) 2.37 -2.44 (m, 2 H) 2.68 (s, 3 H) 2.93 (t, .7=6.78 Hz, 2 H) 3.74 (s, 3 H) 3.90 (s, 3 H) 4.60 (s, 2 H) 5.08 (spt, 7=6.19 Hz, 1 H) 6.80 (s, 1 H) 7.08 – 7.15 (m, 1 H) 7.19 – 7.26 (m, 2 H) 7.52 (d, .7=8.03 Hz, 1 H) 7.94 – 8.01 (m, 2 H) 8.56 (s, 1 H) 8.66 (s, 1 H).[00367] Step 5: Preparation of isopropyl 2-((4-((2-(dimethylamino)ethyl)(methyl)amino)-2- methoxy-5 -(3 -(phenylsulfonyl)propanamido)phenyl)amino)-4-(l -methyl- lH-indol-3- yl)pyrimidine-5-carboxylate.
lnt-5[00368] A mixture of the product of step 4 (1.284 kg, 2.415 mol, 1.0 eq.) and 3- (phenylsulfonyl)propionic acid (0.5528 kg, 2.580 mol, 1.07 eq.) in anhydrous DCM (8.5 L) was cooled to 2 °C, and treated with DIEA (0.310 kg, 2.399 mol, 1.0 eq.). To the reaction mixture was charged over 40 min, 50 % w/w T3P in MeTHF (1.756 kg, 2.759 mol, 1.14 eq.) while maintaining the internal temperature between 0 °C and 8 °C. The mixture was stirred at 0 °C to 5 °C for 15 minutes and then warmed over 30 min to 15 °C then held at 15 °C to 30 °C for 60 min.[00369] The reaction was quenched with water (179 mL). The reaction mixture was stirred at ambient temperature for 30 min then DIEA (439 g) was charged in one portion. The resulting mixture was aged for 15 min, and then treated with 5% aqueous K2CO3 (7.3 L) at 22-25 °C. The organic layer was separated and the aqueous layer back extracted with DCM (6.142 L). The combined organic extract was washed with brine (2 x 5.5 L).[00370] The organic extract was concentrated to 6.5 L, diluted with EtOFl, 200 Proof (14.3 kg), and the mixture concentrated under vacuum (23-25 inch Flg/IT40-60 °C) to a residual volume of 12.8 L.[00371] The residual slurry was treated with EtOFl, 200 Proof (28.8 Kg), and heated to 69 °C to obtain a thin slurry. The reaction mixture was cooled to 15 °C over 2 h, and stored overnight at 15 °C under nitrogen.[00372] The next day, the mixture was cooled to 5 °C, and aged for 30 minutes. The resulting solid was isolated by filtration, washed with EtOFl (2 x 2.16 kg) and dried to give 1.769 kg (100% yield) of the title compound. F1PLC purity 99.85%.‘H NMR (400 MHz, DMSO-i¾ d ppm 1.08 – 1.19 (m, 8 H) 2.15 (s, 6 H) 2.32 (t, J= 5.77 Hz, 2 H) 2.66 – 2.76 (m, 5 H) 2.88 (br t, J= 5.52 Hz, 2 H) 3.48 (qd, J= 7.03, 5.02 Hz, 1 H) 3.60 – 3.69 (m, 2 H) 3.83 (s, 3 H) 3.89 (s, 3 H) 4.40 (t, J=5.02 Hz, 1 H) 5.04 (quin, J=6.27 Hz, 1 H) 7.01 – 7.09 (m, 2 H) 7.22 (t, J= 7.53 Hz, 1 H) 7.52 (d, J= 8.53 Hz, 1 H) 7.67 – 7.82 (m, 4 H) 7.97 (s, 1 H) 7.98 – 8.00 (m, 1 H) 8.14 (s, 1 H) 8.61 – 8.70 (m, 3 H) 10.09 (s, 1 H).[00373] Step 6: Preparation of isopropyl 2-((5-acrylamido-4-((2-(dimethylamino)ethyl) (methyl)amino)-2-methoxyphenyl)amino)-4-(l -methyl- lH-indol-3-yl)pyrimidine-5-carboxylate (Compound (A)).
compound (A)[00374] The product of step 5 (1.600 kg, 2.198 mol, 1.0 equiv.) was dissolved in anhydrous THF (19.5 kg) and was treated at -1 °C to 1 °C with 2M KOSi(CH3)3 in THF (2.72 L, 5.44 mol, 2.47 equiv.). KOSi(CFb)3 was added over 5 minutes, reactor jacket set at -5 °C to 10 °C. 2 M KOSi(CFh)3 solution was prepared by dissolving 871 g of KOSi(CFh)3 technical grade (90%) in 3.056 L of anhydrous TF1F.[00375] The reaction mixture was aged for 60 minutes. Potable water (22 L) was charged to the reaction mixture over 1 10 minutes, while maintaining temperature at 2-7 °C. The resulting suspension was aged at 3-7 °C for 60 minutes; the product was isolated by filtration (the filtration rate during crude product isolation was (1.25 L/min), washed with potable water (2 x 1.6 L) and air dried overnight and then in high vacuum for 12 h at 45 °C to give 1.186 kg of crude title compound (92% yield).‘H NMR (500 MHz, DMSO-i¾ d ppm 1.05 (t, J= 7.09 Hz, 2 H) 1.1 1 (d, J= 6.36 Hz, 6 H) 2.1 1 (s, 6 H) 2.28 (br t, .7=5.38 Hz, 3 H) 2.55 – 2.67 (m, 3 H) 2.69 (s, 3 H) 2.83 (br t, .7=5.38 Hz, 3 H) 3.31 (s, 3 H) 3.36 – 3.51 (m, 2 H) 3.54 – 3.70 (m, 3 H) 3.75 – 3.82 (m, 3 H) 4.33 (t, .7=5.14 Hz, 1 H) 4.99 (dt, 7=12.35, 6.30 Hz, 2 H) 5.75 (s, 1 H) 6.95 – 7.07 (m, 2 H) 7.17 (br t, .7=7.58 Hz, 2 H) 7.48 (d, 7=8.31 Hz, 2 H) 7.62 – 7.71 (m, 3 H) 7.71 – 7.83 (m, 2 H) 7.93 (d, .7=7.83 Hz, 3 H) 8.09 (s, 2 H) 8.53 – 8.67 (m, 3 H) 10.03 (s, 2 H).[00376] Step 7: Preparation of polymorphic Form-I of isopropyl 2-((5-acrylamido-4-((2- (dimethylamino)ethyl) (methyl)amino)-2-methoxyphenyl)amino)-4-(l -methyl- lH-indol-3- yl)pyrimidine-5-carboxylate (Free base Compound (A)).[00377] Method 1 : The crude product of step 6 (1.130 kg) was recrystallized by dissolving it in EtOAc (30.1 kg) at 75 °C, polish filtered (1.2 pm in-line filter), followed by concentration of the filtrate to 14 L of residue (IT during concentration is 58-70 °C). The residual slurry was cooled to 0 °C over 70 minutes and then aged at 0-2 °C for 30 minutes. Upon isolation the product was dried to a constant weight to give 1.007 kg (89% recovery) of the title compound as polymorphic Form-I. Purity (HPLC, a/a %, 99.80%).
Novel inhibitor of programmed cell dealth-1 (PD-1)
CA-170 (also known as AUPM170 or PD-1-IN-1) is a first-in-class, potent and orally available small molecule inhibitor of the immune checkpoint regulatory proteins PD-L1 (programmed cell death ligand-1), PD-L2 and VISTA (V-domain immunoglobulin (Ig) suppressor of T-cell activation (programmed death 1 homolog; PD-1H). CA-170 was discovered by Curis Inc. and has potential antineoplastic activities. CA-170 selectively targets PD-L1 and VISTA, both of which function as negative checkpoint regulators of immune activation. Curis is currently investigating CA-170 for the treatment of advanced solid tumours and lymphomas in patients in a Phase 1 trial (ClinicalTrials.gov Identifier: NCT02812875).
Curis and Aurigene Announce Amendment of Collaboration for the Development and Commercialization of CA-170
– Aurigene to fund and conduct a Phase 2b/3 randomized study of CA-170 in patients with non-squamous non-small cell lung cancer (nsNSCLC) –
– Aurigene to receive Asia rights for CA-170; Curis entitled to royalty payments in Asia –
LEXINGTON, Mass., February 5, 2020 /PRNewswire/ — Curis, Inc. (NASDAQ: CRIS), a biotechnology company focused on the development of innovative therapeutics for the treatment of cancer, today announced that it has entered into an amendment of its collaboration, license and option agreement with Aurigene Discovery Technologies, Ltd. (Aurigene). Under the terms of the amended agreement, Aurigene will fund and conduct a Phase 2b/3 randomized study evaluating CA-170, an orally available, dual inhibitor of VISTA and PDL1, in combination with chemoradiation, in approximately 240 patients with nonsquamous non-small cell lung cancer (nsNSCLC). In turn, Aurigene receives rights to develop and commercialize CA-170 in Asia, in addition to its existing rights in India and Russia, based on the terms of the original agreement. Curis retains U.S., E.U., and rest of world rights to CA-170, and is entitled to receive royalty payments on potential future sales of CA-170 in Asia.
In 2019, Aurigene presented clinical data from a Phase 2a basket study of CA-170 in patients with multiple tumor types, including those with nsNSCLC. In the study, CA-170 demonstrated promising signs of safety and efficacy in nsNSCLC patients compared to various anti-PD-1/PD-L1 antibodies.
“We are pleased to announce this amendment which leverages our partner Aurigene’s expertise and resources to support the clinical advancement of CA-170, as well as maintain our rights to CA-170 outside of Asia,” said James Dentzer, President and Chief Executive Officer of Curis. “Phase 2a data presented at the European Society for Medical Oncology (ESMO) conference last fall supported the potential for CA-170 to serve as a therapeutic option for patients with nsNSCLC. We look forward to working with our partner Aurigene to further explore this opportunity.”
“Despite recent advancements, patients with localized unresectable NSCLC struggle with high rates of recurrence and need for expensive intravenous biologics. The CA-170 data presented at ESMO 2019 from Aurigene’s Phase 2 ASIAD trial showed encouraging results in Clinical Benefit Rate and Prolonged PFS and support its potential to provide clinically meaningful benefit to Stage III and IVa nsNSCLC patients, in combination with chemoradiation and as oral maintenance” said Kumar Prabhash, MD, Professor of Medical Oncology at Tata Memorial Hospital, Mumbai, India.
Murali Ramachandra, PhD, Chief Executive Officer of Aurigene, commented, “Development of CA-170, with its unique dual inhibition of PD-L1 and VISTA, is the result of years of hard-work and commitment by many people, including the patients who participated in the trials, caregivers and physicians, along with the talented teams at Aurigene and Curis. We look forward to further developing CA-170 in nsNSCLC.”
About Curis, Inc.
Curis is a biotechnology company focused on the development of innovative therapeutics for the treatment of cancer, including fimepinostat, which is being investigated in combination with venetoclax in a Phase 1 clinical study in patients with DLBCL. In 2015, Curis entered into a collaboration with Aurigene in the areas of immuno-oncology and precision oncology. As part of this collaboration, Curis has exclusive licenses to oral small molecule antagonists of immune checkpoints including, the VISTA/PDL1 antagonist CA-170, and the TIM3/PDL1 antagonist CA-327, as well as the IRAK4 kinase inhibitor, CA- 4948. CA-4948 is currently undergoing testing in a Phase 1 trial in patients with non-Hodgkin lymphoma. In addition, Curis is engaged in a collaboration with ImmuNext for development of CI-8993, a monoclonal anti-VISTA antibody. Curis is also party to a collaboration with Genentech, a member of the Roche Group, under which Genentech and Roche are commercializing Erivedge® for the treatment of advanced basal cell carcinoma. For more information, visit Curis’ website at http://www.curis.com.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY, NYSE: RDY). Aurigene is focused on precision- oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene currently has several programs from its pipeline in clinical development. Aurigene’s ROR-gamma inverse agonist AUR-101 is currently in phase 2 clinical development under a US FDA IND. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has partnered with many large and mid-pharma companies in the United States and Europe and has 15 programs currently in clinical development. For more information, please visit Aurigene’s website at https://www.aurigene.com/
Curis with the option to exclusively license Aurigene’s orally-available small molecule antagonist of programmed death ligand-1 (PD-L1) in the immuno-oncology field
Addressing immune checkpoint pathways is a well validated strategy to treat human cancers and the ability to target PD-1/PD-L1 and other immune checkpoints with orally available small molecule drugs has the potential to be a distinct and major advancement for patients.
Through its collaboration with Aurigene, Curis is now engaged in the discovery and development of the first ever orally bioavailable, small molecule antagonists that target immune checkpoint receptor-ligand interactions, including PD-1/PD-L1 interactions. In the first half of 2016, Curis expects to file an IND application with the U.S. FDA to initiate clinical testing of CA-170, the first small molecule immune checkpoint antagonist targeting PD-L1 and VISTA. The multi-year collaboration with Aurigene is focused on generation of small molecule antagonists targeting additional checkpoint receptor-ligand interactions and Curis expects to advance additional drug candidates for clinical testing in the coming years. The next immuno-oncology program in the collaboration is currently targeting the immune checkpoints PD-L1 and TIM3.
In November 2015, preclinical data were reported. Data demonstrated tha the drug rescued and sustained activation of T cells functions in culture. CA-170 resulted in anti-tumor activity in multiple syngeneic tumor models including melanoma and colon cancer. Similar data were presented at the 2015 AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Conference in Boston, MA
By August 2015, preclinical data had been reported. Preliminary data demonstrated that in in vitro studies, small molecule PD-L1 antagonists induced effective T cell proliferation and IFN-gamma production by T cells that were specifically suppressed by PD-L1 in culture. The compounds were found to have effects similar to anti-PD1 antibodies in in vivo tumor models
(Oral Small Molecule PD-L1/VISTAAntagonist)
Certain human cancers express a ligand on their cell surface referred to as Programmed-death Ligand 1, or PD-L1, which binds to its cognate receptor, Programmed-death 1, or PD-1, present on the surface of the immune system’s T cells. Cell surface interactions between tumor cells and T cells through PD-L1/PD-1 molecules result in T cell inactivation and hence the inability of the body to mount an effective immune response against the tumor. It has been previously shown that modulation of the PD-1 mediated inhibition of T cells by either anti-PD1 antibodies or anti-PD-L1 antibodies can lead to activation of T cells that result in the observed anti-tumor effects in the tumor tissues. Therapeutic monoclonal antibodies targeting the PD-1/PD-L1 interactions have now been approved by the U.S. FDA for the treatment of certain cancers, and multiple therapeutic monoclonal antibodies targeting PD-1 or PD-L1 are currently in development.
In addition to PD-1/PD-L1 immune regulators, there are several other checkpoint molecules that are involved in the modulation of immune responses to tumor cells1. One such regulator is V-domain Ig suppressor of T-cell activation or VISTA that shares structural homology with PD-L1 and is also a potent suppressor of T cell functions. However, the expression of VISTA is different from that of PD-L1, and appears to be limited to the hematopoietic compartment in tissues such as spleen, lymph nodes and blood as well as in myeloid hematopoietic cells within the tumor microenvironment. Recent animal studies have demonstrated that combined targeting/ blockade of PD-1/PD-L1 interactions and VISTA result in improved anti-tumor responses in certain tumor models, highlighting their distinct and non-redundant functions in regulating the immune response to tumors2.
As part of the collaboration with Aurigene, in October 2015 Curis licensed a first-in-class oral, small molecule antagonist designated as CA-170 that selectively targets PD-L1 and VISTA, both of which function as negative checkpoint regulators of immune activation. CA-170 was selected from the broad PD-1 pathway antagonist program that the companies have been engaged in since the collaboration was established in January 2015. Preclinical data demonstrate that CA-170 can induce effective proliferation and IFN-γ (Interferon-gamma) production (a cytokine that is produced by activated T cells and is a marker of T cell activation) by T cells that are specifically suppressed by PD-L1 or VISTA in culture. In addition, CA-170 also appears to have anti-tumor effects similar to anti-PD-1 or anti-VISTA antibodies in multiple in vivo tumor models and appears to have a good in vivo safety profile. Curis expects to file an IND and initiate clinical testing of CA-170 in patients with advanced tumors during the first half of 2016.
Jan 21, 2015
Curis and Aurigene Announce Collaboration, License and Option Agreement to Discover, Develop and Commercialize Small Molecule Antagonists for Immuno-Oncology and Precision Oncology Targets
— Agreement Provides Curis with Option to Exclusively License Aurigene’s Antagonists for Immuno-Oncology, Including an Antagonist of PD-L1 and Selected Precision Oncology Targets, Including an IRAK4 Kinase Inhibitor —
— Investigational New Drug (IND) Application Filings for Both Initial Collaboration Programs Expected this Year —
— Curis to issue 17.1M shares of its Common Stock as Up-front Consideration —
— Management to Host Conference Call Today at 8:00 a.m. EST —
LEXINGTON, Mass. and BANGALORE, India, Jan. 21, 2015 (GLOBE NEWSWIRE) — Curis, Inc. (Nasdaq:CRIS), a biotechnology company focused on the development and commercialization of innovative drug candidates for the treatment of human cancers, and Aurigene Discovery Technologies Limited, a specialized, discovery stage biotechnology company developing novel therapies to treat cancer and inflammatory diseases, today announced that they have entered into an exclusive collaboration agreement focused on immuno-oncology and selected precision oncology targets. The collaboration provides for inclusion of multiple programs, with Curis having the option to exclusively license compounds once a development candidate is nominated within each respective program. The partnership draws from each company’s respective areas of expertise, with Aurigene having the responsibility for conducting all discovery and preclinical activities, including IND-enabling studies and providing Phase 1 clinical trial supply, and Curis having responsibility for all clinical development, regulatory and commercialization efforts worldwide, excluding India and Russia, for each program for which it exercises an option to obtain a license.
The first two programs under the collaboration are an orally-available small molecule antagonist of programmed death ligand-1 (PD-L1) in the immuno-oncology field and an orally-available small molecule inhibitor of Interleukin-1 receptor-associated kinase 4 (IRAK4) in the precision oncology field. Curis expects to exercise its option to obtain exclusive licenses to both programs and file IND applications for a development candidate from each in 2015.
“We are thrilled to partner with Aurigene in seeking to discover, develop and commercialize small molecule drug candidates generated from Aurigene’s novel technology and we believe that this collaboration represents a true transformation for Curis that positions the company for continued growth in the development and eventual commercialization of cancer drugs,” said Ali Fattaey, Ph.D., President and Chief Executive Officer of Curis. “The multi-year nature of our collaboration means that the parties have the potential to generate a steady pipeline of novel drug candidates in the coming years. Addressing immune checkpoint pathways is now a well validated strategy to treat human cancers and the ability to target PD-1/PD-L1 and other immune checkpoints with orally available small molecule drugs has the potential to be a distinct and major advancement for patients. Recent studies have also shown that alterations of the MYD88 gene lead to dysregulation of its downstream target IRAK4 in a number of hematologic malignancies, including Waldenström’s Macroglobulinemia and a subset of diffuse large B-cell lymphomas, making IRAK4 an attractive target for the treatment of these cancers. We look forward to advancing these programs into clinical development later this year.”
Dr. Fattaey continued, “Aurigene has a long and well-established track record of generating targeted small molecule drug candidates with bio-pharmaceutical collaborators and we have significantly expanded our drug development capabilities as we advance our proprietary drug candidates in currently ongoing clinical studies. We believe that we are well-positioned to advance compounds from this collaboration into clinical development.”
CSN Murthy, Chief Executive Officer of Aurigene, said, “We are excited to enter into this exclusive collaboration with Curis under which we intend to discover and develop a number of drug candidates from our chemistry innovations in the most exciting fields of cancer therapy. This unique collaboration is an opportunity for Aurigene to participate in advancing our discoveries into clinical development and beyond, and mutually align interests as provided for in our agreement. Our scientists at Aurigene have established a novel strategy to address immune checkpoint targets using small molecule chemical approaches, and have discovered a number of candidates that modulate these checkpoint pathways, including PD-1/PD-L1. We have established a large panel of preclinical tumor models in immunocompetent mice and can show significant in vivo anti-tumor activity using our small molecule PD-L1 antagonists. We are also in the late stages of selecting a candidate that is a potent and selective inhibitor of the IRAK4 kinase, demonstrating excellent in vivo activity in preclinical tumor models.”
In connection with the transaction, Curis has issued to Aurigene approximately 17.1 million shares of its common stock, or 19.9% of its outstanding common stock immediately prior to the transaction, in partial consideration for the rights granted to Curis under the collaboration agreement. The shares issued to Aurigene are subject to a lock-up agreement until January 18, 2017, with a portion of the shares being released from the lock-up in four equal bi-annual installments between now and that date.
The agreement provides that the parties will collaborate exclusively in immuno-oncology for an initial period of approximately two years, with the option for Curis to extend the broad immuno-oncology exclusivity.
In addition Curis has agreed to make payments to Aurigene as follows:
for the first two programs: up to $52.5 million per program, including $42.5 million per program for approval and commercial milestones, plus specified approval milestone payments for additional indications, if any;
for the third and fourth programs: up to $50 million per program, including $42.5 million per program for approval and commercial milestones, plus specified approval milestone payments for additional indications, if any; and
for any program thereafter: up to $140.5 million per program, including $87.5 million per program in approval and commercial milestones, plus specified approval milestone payments for additional indications, if any.
Curis has agreed to pay Aurigene royalties on any net sales ranging from high single digits to 10% in territories where it successfully commercializes products and will also share in amounts that it receives from sublicensees depending upon the stage of development of the respective molecule. About Immune Checkpoint Modulation and Programmed Death 1 Pathway
Modulation of immune checkpoint pathways has emerged as a highly promising therapeutic approach in a wide range of human cancers. Immune checkpoints are critical for the maintenance of self-tolerance as well as for the protection of tissues from excessive immune response generated during infections. However, cancer cells have the ability to modulate certain immune checkpoint pathways as a mechanism to evade the immune system. Certain immune checkpoint receptors or ligands are expressed by various cancer cells, targeting of which may be an effective strategy for generating anti-tumor activity. Some immune-checkpoint modulators, such as programmed death 1 (PD-1) protein, specifically regulate immune cell effector functions within tissues. One of the mechanisms by which tumor cells block anti-tumor immune responses in the tumor microenvironment is by upregulating ligands for PD-1, such as PD-L1. Hence, targeting of PD-1 and/or PD-L1 has been shown to lead to the generation of effective anti-tumor responses. About Curis, Inc.
Curis is a biotechnology company focused on the development and commercialization of novel drug candidates for the treatment of human cancers. Curis’ pipeline of drug candidates includes CUDC-907, a dual HDAC and PI3K inhibitor, CUDC-427, a small molecule antagonist of IAP proteins, and Debio 0932, an oral HSP90 inhibitor. Curis is also engaged in a collaboration with Genentech, a member of the Roche Group, under which Genentech and Roche are developing and commercializing Erivedge®, the first and only FDA-approved medicine for the treatment of advanced basal cell carcinoma. For more information, visit Curis’ website at www.curis.com.
About Aurigene
Aurigene is a specialized, discovery stage biotechnology company, developing novel and best-in-class therapies to treat cancer and inflammatory diseases. Aurigene’s Programmed Death pathway program is the first of several immune checkpoint programs that are at different stages of discovery and preclinical development. Aurigene has partnered with several large- and mid-pharma companies in the United States and Europe and has delivered multiple clinical compounds through these partnerships. With over 500 scientists, Aurigene has collaborated with 6 of the top 10 pharma companies. Aurigene is an independent, wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (NYSE:RDY). For more information, please visit Aurigene’s website at http://aurigene.com/.
POSTER
WO2011161699, WO2012/168944, WO2013144704 and WO2013132317 report peptides or peptidomimetic compounds which are capable of suppressing and/or inhibiting the programmed cell death 1 (PD1) signaling pathway.
The compound was synthesised using similar procedure as depicted in Example 2 for synthesising compound 2 using instead of H-Ser(‘Bu)-0’Bu (in synthesis of compound 2b) to yield 0.35 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 361.2 (M+H)+, HPLC: tR = 12.19 min.
RP-12146 is an oral poly (ADP-ribose) polymerase (PARP) inhibitor in phase I clinical development at Rhizen Pharmaceuticals for the treatment of adult patients with locally advanced or metastatic solid tumors.
Solid TumorExtensive-stage Small-cell Lung CancerLocally Advanced Breast CancerMetastatic Breast CancerPlatinum-sensitive Ovarian CancerPlatinum-Sensitive Fallopian Tube CarcinomaPlatinum-Sensitive Peritoneal Cancer
Poly(ADP-ribose) polymerase (PARP) defines a family of 17 enzymes that cleaves NAD+ to nicotinamide and ADP-ribose to form long and branched (ADP-ribose) polymers on glutamic acid residues of a number of target proteins, including PARP itself. The addition of negatively charged polymers profoundly alters the properties and functions of the acceptor proteins. Poly(ADP-ribosyl)ation is involved in the regulation of many cellular processes, such as DNA repair, gene transcription, cell cycle progression, cell death, chromatin functions and genomic stability. These functions have been mainly attributed to PARP-1 that is regarded as the best characterized member of the PARP family. However, the identification of novel genes encoding PARPs, together with the characterization of their structure and subcellular localization, have disclosed different roles for poly(ADP-ribosyl)ation in cells, including telomere replication and cellular transport.
Recently, poly(ADP-ribose) binding sites have been identified in many DNA damage checkpoint proteins, such as tumor suppressor p53, cyclin-dependent kinase inhibitor p21Cip1/waf1, DNA damage recognition factors (i.e., the nucleotide excision repair xeroderma pigmentosum group A complementing protein and the mismatch repair protein MSH6), base excision repair (BER) proteins (i.e. DNA ligase III, X-ray repair cross-complementing 1, and XRCC1), DNA-dependent protein kinase (DNA-PK), cell death and survival regulators (i.e.,
NF-kB, inducible nitric oxide synthase, and telomerase). These findings suggest that the different components of the PARP family might be involved in the DNA damage signal network, thus regulating protein-protein and protein-DNA interactions and, consequently, different types of cellular responses to genotoxic stress. In addition to its involvement in BER and single strand breaks (SSB) repair, PARP-1 appears to aid in the non-homologous end-joining (NHEJ) and homologous recombination (HR) pathways of double strand breaks (DSB) repair. See Lucio Tentori et al., Pharmacological Research, Vol. 45, No. 2, 2002, page 73-85.
PARP inhibition might be a useful therapeutic strategy not only for the treatment of BRCA mutations but also for the treatment of a wider range of tumors bearing a variety of deficiencies in the HR pathway. Further, the existing clinical data (e.g., Csaba Szabo et al., British Journal of Pharmacology (2018) 175: 192-222) also indicate that stroke, traumatic brain injury, circulatory shock and acute myocardial infarction are some of the indications where PARP activation has been demonstrated to contribute to tissue necrosis and inflammatory responses.
As of now, four PARP inhibitors, namely olaparib, talazoparib, niraparib, and rucaparib have been approved for human use by regulatory authorities around the world.
Patent literature related to PARP inhibitors includes International Publication Nos. WO 2000/42040, WO 2001/016136, WO 2002/036576, WO 2002/090334, WO2003/093261, WO 2003/106430, WO 2004/080976, WO 2004/087713, WO 2005/012305, WO 2005/012524, WO 2005/012305, WO 2005/012524, WO 2005/053662, W02006/033003, W02006/033007, WO 2006/033006, WO 2006/021801, WO 2006/067472, WO 2007/144637, WO 2007/144639, WO 2007/144652, WO 2008/047082, WO 2008/114114, WO 2009/050469, WO 2011/098971, WO 2015/108986, WO 2016/028689, WO 2016/165650, WO 2017/153958, WO 2017/191562, WO 2017/123156, WO 2017/140283, WO 2018/197463, WO 2018/038680 and WO 2018/108152, each of which is incorporated herein by reference in its entirety for all purposes.
There still remains an unmet need for new PARP inhibitors for the treatment of various diseases and disorders associated with cell proliferation, such as cancer.
Abstract 1233: Preclinical profile of RP12146, a novel, selective, and potent small molecule inhibitor of PARP1/2
Srikant Viswanadha, Satyanarayana Eleswarapu, Kondababu Rasamsetti, Debnath Bhuniya, Gayatriswaroop Merikapudi, Sridhar Veeraraghavan and Swaroop VakkalankaProceedings: AACR Annual Meeting 2021; April 10-15, 2021 and May 17-21, 2021; Philadelphia, PA
Abstract
Background: Poly (ADP-ribose) polymerase (PARP) activity involves synthesis of Poly-ADP ribose (PAR) polymers that recruit host DNA repair proteins leading to correction of DNA damage and maintenance of cell viability. Upon combining with DNA damaging cytotoxic agents, PARP inhibitors have been reported to demonstrate chemo- and radio-potentiation albeit with incidences of myelosuppression. A need therefore exists for the development selective PARP1/2 inhibitors with a high therapeutic window to fully exploit their potential as a single agent or in combination with established therapy across various tumor types. Additionally, with the emerging concept of ‘synthetic lethality’, the applicability PARP inhibitors can be expanded to cancers beyond the well-defined BRCA defects. Herein, we describe the preclinical profile of RP12146, a novel and selective small molecule inhibitor of PARP1 and PARP2.
Methods: Enzymatic potency was evaluated using a PARP Chemiluminescent Activity Assay Kit (BPS biosciences). Cell growth was determined following incubation with RP12146 in BRCA1 mutant and wild-type cell lines across indications. Apoptosis was evaluated following incubation of cell lines with compound for 120 h, subsequent staining with Annexin-V-PE and 7-AAD, and analysis by flow cytometry. For cell cycle, cells were incubated with compound for 72 h, and stained with Propidium Iodide prior to analysis by flow cytometry. Expression of downstream PAR, PARP-trapping, phospho-γH2AX and cleaved PARP expression were determined in UWB1.289 (BRCA1 null) cells by Western blotting. Anti-tumor potential of RP12146 was tested in OVCAR-3 Xenograft model. Pharmacokinetic properties of the molecule were also evaluated. Results: RP12146 demonstrated equipotent inhibition of PARP1 (0.6 nM) and PARP2 (0.5 nM) with several fold selectivity over the other members of the PARP family. Compound caused a dose-dependent growth inhibition of both BRCA mutant and non-mutant cancer cell lines with GI50 in the range of 0.04 µM to 9.6 µM. Incubation of UWB1.289 cells with RP12146 caused a G2/M arrest with a corresponding dose-dependent increase in the percent of apoptotic cells. Expression of PAR was inhibited by 86% at 10 nM with a 2.3-fold increase in PARP-trapping observed at 100 nM in presence of RP12146. A four-fold increase in phospho-γH2AX and > 2-fold increase in cleaved PARP expression was observed at 3 µM of the compound. RP12146 exhibited anti-tumor potential with TGI of 28% as a single agent in OVCAR-3 xenograft model. Efficay was superior compared to Olaparib tested at an equivalent dose. Pharmacokinetic studies in rodents indicated high bioavailability with favorable plasma concentrations relevant for efficacy
Conclusions: Data demonstrate the therapeutic potential of RP12146 in BRCA mutant tumors. Testing in patients is planned in H1 2021.
Citation Format: Srikant Viswanadha, Satyanarayana Eleswarapu, Kondababu Rasamsetti, Debnath Bhuniya, Gayatriswaroop Merikapudi, Sridhar Veeraraghavan, Swaroop Vakkalanka. Preclinical profile of RP12146, a novel, selective, and potent small molecule inhibitor of PARP1/2 [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2021; 2021 Apr 10-15 and May 17-21. Philadelphia (PA): AACR; Cancer Res 2021;81(13_Suppl):Abstract nr 1233.
Rhizen Pharmaceuticals AG Announces First Patient Dosing in a Phase I/Ib Study of Its Novel PARP Inhibitor (RP12146) in Patients With Advanced Solid Tumors
RHIZEN’S PARP INHIBITOR EFFORTS ARE PART OF A LARGER DDR PLATFORM THAT ALSO INCLUDES AN EARLY STAGE POLθ-DIRECTED PROGRAM; PLATFORM ENABLES PROPRIETARY IN-HOUSE COMBINATIONS
Rhizen Pharma commences dosing in a phase I/Ib trial to evaluate its novel PARP inhibitor (RP12146) in patients with advanced cancers.
Rhizen indicated that RP12146 has comparable preclinical activity vis-à-vis approved PARP inhibitors and shows improved preclinical safety that it expects will translate in the clinic.
The two-part multi-center phase I/Ib study is being conducted in Europe and is designed to initially determine safety, tolerability and MTD/RP2D of RP12146 and to subsequently assess its anti-tumor activity in expansion cohorts with HRR mutation-enriched ES-SCLC, ovarian and breast cancer patients.
RP12146 is part of a larger DDR platform at Rhizen that includes a preclinical-stage Polθ inhibitor program; the DDR platform enables novel, proprietary, in-house combinations
November 01, 2021 07:24 AM Eastern Daylight Time
BASEL, Switzerland–(BUSINESS WIRE)–Rhizen Pharmaceuticals AG (Rhizen), a Switzerland-based privately held, clinical-stage oncology & inflammation-focused biopharmaceutical company, announced today that it has commenced dosing in a multi-center, phase I/Ib trial to evaluate its novel poly (ADP-ribose) polymerase (PARP) inhibitor (RP12146) in patients with advanced solid tumors. This two-part multi-center phase I/Ib study is being conducted in Europe and has been designed to initially determine safety, tolerability, maximum tolerated dose (MTD), and/or recommended phase II dose (RP2D) of RP12146 and to subsequently assess its anti-tumor activity in expansion cohorts with HRR mutation-enriched ES-SCLC, ovarian and breast cancer patients.
“Our PARP program is foundational for our DDR platform efforts and will be the backbone for several novel proprietary combinations that we hope to bring into development going forward.”
Rhizen indicated that RP12146 has shown preclinical activity and efficacy comparable to the approved PARP inhibitor Olaparib, and shows improved safety as seen in the preclinical IND-enabling toxicology studies; an advantage that Rhizen hopes will translate in the clinical studies. Rhizen also announced that its PARP program is part of a larger DNA Damage Response (DDR) platform effort, which includes a preclinical-stage polymerase theta (Polθ) inhibitor program. Rhizen expects the platform to enable novel proprietary combinations of its PARP and Polθ assets given the mechanistic synergy and opportunity across PARP resistant/refractory settings.
“PARP inhibitors are a great success story in the DNA damage response area, but they are not without safety concerns that have limited realization of their full potential. Although our novel PARP inhibitor is competing in a crowded space, we expect its superior preclinical safety to translate into the clinic which will differentiate our program and allow us to extend its application beyond the current landscape of approved indications and combinations”, said Swaroop Vakkalanka, Founder & CEO of Rhizen Pharma. Swaroop also added that “Our PARP program is foundational for our DDR platform efforts and will be the backbone for several novel proprietary combinations that we hope to bring into development going forward.”
About Rhizen Pharmaceuticals AG.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel oncology & inflammation therapeutics. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways.
Rhizen has proven expertise in the PI3K modulator space with the discovery of our first PI3Kδ & CK1ε asset Umbralisib, that has been successfully developed & commercialized in MZL & FL by our licensing partner TG Therapeutics (TGTX) in USA. Beyond this, Rhizen has a deep oncology & inflammation pipeline spanning discovery to phase II clinical development stages.
Rhizen is headquartered in Basel, Switzerland.
REF
Safety, Pharmacokinetics and Anti-tumor Activity of RP12146, a PARP Inhibitor, in Patients With Locally Advanced or Metastatic Solid Tumors….https://clinicaltrials.gov/ct2/show/NCT05002868
TNO155 is a potent selective and orally active allosteric inhibitor of wild-type SHP2 (IC50=0.011 µM). TNO155 has the potential for the study of RTK-dependent malignancies, especially advanced solid tumors.
Originator Novartis
Developer Mirati Therapeutics; Novartis
Class Antineoplastics
Mechanism of ActionProtein tyrosine phosphatase non receptor antagonists
Phase I/IISolid tumours
Phase IColorectal cancer
11 Jul 2021Phase I trial in Solid tumours is still ongoing in USA, Canada, Japan, South Korea, Netherlands, Singapore, Spain, Taiwan (NCT03114319)
04 Jun 2021Efficacy, safety and pharmacokinetics data from phase I trial in Solid tumours presented at 57th Annual Meeting of the American Society of Clinical Oncology (ASCO-2021)
08 Jan 2021Novartis plans a phase Ib/II trial for Solid tumours (Combination therapy, Inoperable/Unresectable, Late-stage disease, Metastatic disease, Second-line therapy or greater) in February 2021 (NCT04699188)
CLIP
Combinations with Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase Signaling
Results: In EGFR-mutant lung cancer models, combination benefit of TNO155 and the EGFRi nazartinib was observed, coincident with sustained ERK inhibition. In BRAFV600E colorectal cancer models, TNO155 synergized with BRAF plus MEK inhibitors by blocking ERK feedback activation by different RTKs. In KRASG12C cancer cells, TNO155 effectively blocked the feedback activation of wild-type KRAS or other RAS isoforms induced by KRASG12Ci and greatly enhanced efficacy. In addition, TNO155 and the CDK4/6 inhibitor ribociclib showed combination benefit in a large panel of lung and colorectal cancer patient–derived xenografts, including those with KRAS mutations. Finally, TNO155 effectively inhibited RAS activation by colony-stimulating factor 1 receptor, which is critical for the maturation of immunosuppressive tumor-associated macrophages, and showed combination activity with anti–PD-1 antibody.
Conclusions: Our findings suggest TNO155 is an effective agent for blocking both tumor-promoting and immune-suppressive RTK signaling in RTK- and MAPK-driven cancers and their tumor microenvironment. Our data provide the rationale for evaluating these combinations clinically.
(3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine, which has the formula I,
WO/2015/107495 A1 describes a method for the manufacture of the compound of the formula I which can be characterized by the following reaction scheme 1:
Scheme 1:
[0008] The last compound resulting from step g above was then reacted as in the following scheme 2:
Scheme 2:
[0009] Thus the compound of formula I is obtained (last compound in the scheme 2, above). The synthesis requires at least the 9 steps shown and is rather appropriate for synthesis in laboratory amounts.
Scheme 1A:
[0016] Therefore, the process, though readily feasible on a laboratory scale, is not ideal for manufacture at a large scale.
[0017] The compound added in reaction b in Scheme 2 is obtained in WO
2015/107495 A1 as “Intermediate 10” follows:
Scheme 3:
[0018] An issue here is the relatively low yield of the amine resulting from reaction a in
Scheme 3.
[0019] In addition, while WO 2015/107495 A1 generically mentions that pharmaceutically acceptable salts of the compound of the formula I may be obtainable, no concrete reason for obtaining such salts and no specific examples of salts are described.
[0020] In addition, given the many potentially salt forming groups in formula I, it is not clear whether any salts with a clear stoichiometry can be formed at all.
Example 1
Method of synthesis of the compound of the formula I ((3S,4S)-8-(6-amino-5-((2-amino-3- chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine):
The overall synthesis can be described by the following Reaction Scheme A:
Scheme A:
Step a
[00293] To a solution of A1 (10.4 kg, 100 mol, 1.0 Eq) in CH2Cl2 (50 L) was added imidazole (8.16 kg, 120 mol, 1.2eq) and TBSCl (18 kg, 120 mol, 1.2 Eq) at 0 °C. After addition, the mixture was stirred at 0°C for 4 h . GC showed the reaction was finished. (A1/ (A1 + A2) < 1%). The reaction mixture was quenched with saturated NaHCO3 (14L) at 0-5°C. Phases were separated. The organic phase was washed with brine (14L). The organic layer was dried over Na2SO4, concentrated under vacuum at 40-45°C to afford A2 (23.3 kg, assay 88%, yield 94%) which was used for the next step directly. 1H NMR (400 MHz, CDC13) δ = 4.35 (d, J= 8.8 Hz, 1H), 3.74 (s, 3H), 2.48 (s, J= 8.8
Hz, 3H), 0.93 (s, 9H), 0.09 (s, 6H).
Step b
[00294] To a solution of A2 (7.5 kg, 34.3 mol, 1.0 Eq) and N,O-dimethylhydroxylamine hydrochloride (6.69 kg, 68.6mol, 2.0 Eq) in THF (20 L) was added drop-wise a solution
of chloro(isopropyl)magnesium (2 M, 51.45 L, 3.5 Eq) at 0 °C under N2 over 5-6 h. After addition, the reaction mixture was stirred at 0 °C for 1h, GC showed the reaction was finished (A2/(A2+A3) < 2 %). The mixture was quenched with NH4Cl (25 L) slowly by keeping the temperature at 0-5°C. After addition, the reaction mixture was stirred for 30min. Phase was separated. The aqueous layer was extracted with EA(2 x 20 L). The combined organic phase was washed with brine (25L), dried over Na2SO4, concentrated to give A3(9.4 kg, assay 86%, yield 95%) which was used for the next step directly. 1HNMR (400 MHz, CDCl3) δ = 4.67 (m, J= 6.6 Hz, 1H), 3.70 (s, 3H), 3.21 (s, 3H), 3.17 (d, 3H)2.48 (s , J= 6.6 Hz, 3H), 0.90 (s, 9H), 0.10 (s, 3H), 0.08 (s, 3H).
Step c
[00295] To a solution of A3 (7.1 kg, assay 86%, 24.65 mol, 1.0 Eq) in DCM (30 L) was added dropwise a solution of LiAlH4 (2.4 M, 11.3 L, 1.1 Eq) at -70 °C under N2. Then the reaction mixture was stirred at -70 °C for 3h, and TLC showed the reaction was finished (PSC-1). The mixture was warmed to 0 °C, and then quenched with sat. potassium sodium tartrate (35 L) at 0 °C. After addition, DCM (20L) was added and stirred for 2h at 20-25°C. Phases were separated. The aqueous layer was extracted with DCM (25 L). The combined organic phase was charged with sat. citric acid (45L) and stirred at 0°C for 8h. Phase was separated. The organic phase was washed with NaHCO3 (25L), brine (25 L), dried over Na2SO4, and the solvent was removed under vacuum at 25-30°C. n-Heptane (10 L) was added to the residue and concentrated under vacuum at 30-35°C. n-Heptane (10 L) was added to the residue again and concentrated under vacuum at 30-35°C to give A4 (4.2 kg, assay
60%, yield 54%) which was used for the next step directly.
Step d
[00296] To a solution of diisopropylamine (3.06 kg, 30.3 mol, 1.5 eq) in THF (20 L) cooled to approximately -10°C was added 2.5 M n-BuLi (12.12 L, 30.3 mol, 1.5 eq) under N2. The resulting mixture was stirred at approximately -10 °C for 30min, then a solution of A5 (5.2 kg, 20.20 mol, 1.0eq) in THF (10 L) was added slowly. After addition, the reaction mixture was stirred at -10°C for 30 min, and then cooled to -50°C. A4 (4.18 kg, 22.22 mol, 1.1eq) was added dropwise. After addition, the reaction mixture was stirred at -50°C for 30 min. The mixture was quenched with saturated aqueous NH4Cl (30L) and water (10L) at -50°C. The reaction mixture was warmed to 20-25°C. Phase was separated. The aqueous phase was extracted with EA (3 x 20 L). All organic phases were combined and washed with brine(20L), then concentrated to a yellow oil which was purified by column (silica gel, 100-200 mesh, eluted with n-heptane:EA from 50:1 to 10:1) to give A6 (5.5 kg, assay 90 %, yield 55%) as pale yellow oil. 1H NMR (400 MHz, CDCl3) δ = 4.35-4.15 (m, 2H), 3.95-3.74 (m, 3H), 3.52 (m, 2H), 2.67(m, 2H), 2.12-1.98 (m, 2H), 1.75-1.52 (m, 4H), 1.49 (s, 9H), 1.35-1.10 (m, 6H), 0.98 (s,
9H), 0.02 (s, 6H).
Step e
[00297] To a solution of A6 (11.4 kg, 25.58 mol, 1.0eq) in THF (60 L) was added LiBH4
(836 g, 38.37 mol, 1.5eq) in portions at 5-10 °C, and the reaction mixture was stirred at 20-25 °C for 18 h. HPLC showed the reaction was finished (A6/(A6+A7)<2%). The mixture was cooled to l0°C and slowly quenched with saturated NaHCO3 solution (15 L) and water (25L) with vigorously stirring. After gas formation stopped, vacuum filtration was applied to remove solids. The solid was washed with EA (2 x 15 L). Phase was separated; the aqueous phase was extracted with EA (3 x15L). All organic phases were combined and washed with brine (15L), and concentrated to obtain crude A7 (13.8 kg, assay 58%, yield 77%) which was used for the next step directly.
Step f
[00298] To a solution of A7 (8 kg, 19.82 mol, 1.0 eq) in THF (40 L) under nitrogen atmosphere was added TsCl (5.28 kg, 27.75 mol, 1.4 eq) at 10-15°C. After addition, the mixture was cooled to 0 °C, and 1M LiHMDS (29.7 L, 29.73 mol, 1.5 eq) was added dropwise during 2h. After addition, the mixture was stirred at 0°C for 3h. HPLC showed the reaction was finished (PSC-1 A7/ (A7+A8)<7%). TBAF (20.72 kg, 65.67 mol, 3.3 eq) was added into the mixture at 0 °C and the reaction mixture was stirred at 25-30 °C for 48h. HPLC showed the reaction was finished ( PSC-2, A9-intermedaite/(A9-intermediate+A9) < 2%). The mixture was quenched with saturated aqueous sodium bicarbonate solution (32L) and stirred for 30min at 0 °C. Phase was separated, and the aqueous phase was extracted with EA (3 x 20 L). The combined organic phase was washed with brine(20 L), dried over Na2SO4, and concentrated to a yellow oil which was purified by column (eluted with n-heptane:EA from 10:1 to 1:1) to give A9 (4.42 kg, assay 90%, yield 74 %) as pale yellow solid.
Step g
[00299] To a solution of A9 (4.0 kg, 14.74 mol, 1.0 eq) in DCM (40 L) cooled on an ice-bath was added DMP (9.36 kg, 23.58mol, 1.6eq) in portions, and it resulted in a suspension. After addition, the mixture stirred for 4 hours at 20-25°C. HPLC showed the reaction was finished (A9/(A9+A10)<2%). DCM (30L) was added at 0°C. After addition, the mixture was quenched with saturated aqueous Na2SO3 (20 L). The mixture was stirred for 30min at 0 °C, filtered and the white solid was washed with DCM (2 x15L). Phase was separated, and the organic phase was cooled to 0°C, to which was added saturated aqueous NaHCO3 (20L) and stirred for 1h. Phase was separated, and the organic phase was washed with brine(25L), dried over Na2SO4, and concentrated to a yellow oil which was purified by column (eluted with n-heptane:EA from 50:1 to 10:1) to give A10 (3.70 kg, assay 88%, ee value 95.3%, yield 82%) as white solid. 1H NMR (400 MHz, DMSO-d6) δ = 4.20 (d, J = 8.0 Hz,
[00300] To a solution of A10 (4.60 kg, 17.08 mol, 1.0 eq) in THF (40 L) was added
Ti(OEt)4 (15.58 kg, 68.32 mol, 4.0 eq) and (R)-t-Butyl sulfmamide (4.14 kg, 34.16 mol, 2.0 eq) at 25 °C. After addition, the mixture was heated to 70°C and stirred for 20h. HPLC showed the reaction was finished (PSC-l, A10/(A10+A12)<4%). The mixture was cooled to -30— 40°C, and MeOH (4 L) was added dropwise within 30 min and stirred for 1 h. 2M L1BH4 (8.1 L) solution was added dropwise to the reaction mixture at -40- -50°C and stirred for 1h. HPLC indicated all of imine was consumed (PSC-2, A12/(A12+A13)<1%). The mixture was warmed to -30 °C and stirred for 1h, then warmed to 0 °C within 2 h and stirred for 1h, then warmed to 20-25 °C and stirred for 30min. IP AC ( 25L) was added to above mixture, NaHCO3(5L) was added dropwise in about 1h at 25 °C and stirred for 30 min. The mixture was filtered under vacuum and the cake was washed with IP AC (8 x15L). The combined organic phase was washed with brine (25L), then evaporated under vacuum to get a solution of A13
(about 28kg) which was used for next step.
Step i
[00301] To a mixture of A13 in IPAC (about 28 kg, 17.08 mol, 1.0 eq) was added dropwise
4M HCl/IPA (8.54 L, 34.16 mol, 2.0 eq) at -5 °C and stirred for 5h at -5 °C. HPLC showed that A13 was consumed completely (A13/(A14+A13)<1%). MTBE (25 L) was added to above mixture within
30 min and stirred for 30 min at -5 °C .The solid was collected by vacuum filtration. The cake was washed with MTBE (2 x 2.5 L). The wet cake was used for next step directly.
Step j
[00302] The wet solid A14 (from 9.2 kg A10) was stirred in MTBE(76 L) at 25°C, then the
16% NaOH (9.84 kg) solution was added dropwise to the MTBE suspension while maintaining IT<10ºC. After addition, the mixture was stirred for 15 min and all solids were dissolved at 0°C. The organic phase was separated, and the aqueous phase was extracted with MTBE (2 x 20L). The combined organic phase was washed with brine (10 L) and evaporated under vacuum to remove all MTBE. ACN (24 L) was added to above residue, and the mixture was evaporated under vacuum to remove the organic solvents and yielded a crude A15 (5.42 kg, qnmr 90%, 18.04 mol, 1.0 eq). ACN (34.68 kg) was added to above residue and stirred for 10 min at 65°C. A solution of (-)-O-acetyl-D-mandelic acid (3.15kg,16.2 mol, 0.9 eq) in ACN(11.6 kg) was added drop-wise to the mixture (firstly added 1/3, stirred for 0.5 h, then added the others) over 3h. The mixture was stirred for 1 h at 65°C, then cooled to 25°C over 4h and stirred for l2h at 25°C . The solid was collected by vacuum filtration, and the cake was washed with pre-cooled ACN (2 x15kg) (PSC-1) and dried under vacuum to give
A16 (7.36 kg, yield 46% from A10 to A16). 1H NMR (400 MHz, DMSO-d6) δ = 7.43-7.29 (m, 5H),
[00303] To a solution of A16 (15 g) in MeOH (90 mL) was added dropwise 5N HC1/IPA
(45 mL) at room temperature within 15 minutes. After the addition, the mixture was stirred for 6 hours.
IP AC (180 mL) was added dropwise to above mixture within 1h at room temperature. The resulting mixture was stirred for another 30 minutes before it was cooled to 0-5 °C. The mixture was stirred at 0- 5 °C for another 2h and the precipitants were collected by filtration. The cake was washed with (45*2 mL) IP AC, dried under vacuum at 60 °C overnight to afford the product as a white solid. 1H NMR (400
[00304] To a mixture of A17 (10 g) and Z17a (9.5 g) in DMAC (60 mL) was added K2CO3
(22.5 g) and H2O (40 mL) at room temperature. The mixture was degassed with nitrogen and stirred at
90 °C overnight. The mixture was cooled to room temperature, diluted with Me-THF (500 mL) and
H2O (280 mL). The organic phase was separated and the aqueous phase was extracted with Me-THF
(300 mL*2). The combined organic phases were washed with brine (200 mL*3), concentrated under
vacuum to remove most of the solvent. The residue was diluted with IPA (60 mL) and H2O (20 mL), stirred at 50 °C for 1h, cooled to 5 °C within 3h, stirred at this temperature for 1h. The solid was collected by vacuum filtration, dried under vacuum to afford the product as a yellow solid (l2g,
Formation of the succinate salt of the compound of the formula I:
[00305] The reaction is summarized by the following Reaction Scheme:
[00306] To a mixture of A18 (10 g) in MeOH (76 g) and H2O (24 g) was added succinic acid (2.94 g) at room temperature. The mixture was heated to 50 °C and stirred for 30 minutes to dissolve all solid. The solution was added to IPA (190 mL) at 60-65 °C. The resulting mixture was stirred at 60 °C >5 hours, cooled to -15 °C within 5 hours and stirred at this temperature >4 hours. The solid was collected by vacuum filtration, dried under vacuum to afford the product as an off-white solid(l0.8 g, 82.8%). 1H NMR (400 MHz, DMSO-d6)δ = 7.64 (d, J= 6.2 Hz, 1H), 7.63 (s, 1H), 6.26 (s, 2H), 6.16 (s, 2H), 5.74 (d, J= 5.3 Hz, 1H), 4.12 – 4.02 (m, 1H), 3.90 – 3.78 (m, 2H), 3.67 (d, J= 8.4 Hz, 1H), 3.49 (d, J= 8.4 Hz, 1H), 3.33 (s, 2H), 2.91 (d, J= 5.1 Hz, 1H), 2.34 (s, 4H), 1.71 – 1.60 (m, 4H), 1.13 (d, J = 6.5 Hz, 3H).
[00307] In a special variant, the reaction follows the following Reaction Scheme, also including an optional milling to yield the final product:
Example 3
Formation of the intermediate Z17a (3-((2-amino-3-chloropyridin-4-yl)thio)-6-chloropyrazin-2- amine). Variant 1:
[00308] The compound Z17a was obtained by reaction according to the following Reaction
Scheme:
[00309] In detail, the synthesis of Compound Z17a was carried out as follows:
Step a
[00310] Under nitrogen atmosphere, n-BuLi (2.5M, 7.6 L) was added dropwise to a solution of 3-chloro-2-fluoropyridine (2 kg) in THF (15 L) at -78°C. Then the resultant mixture was stirred for 1h. Then a solution of I2 (4.82 kg) in THF (6 L) was added dropwise. After addition, the reaction mixture was stirred for 30 min, and then quenched with sat. Na2SO3 (10 L), and warmed to 20- 25°C. Phase was separated. The aqueous phase was extracted with EA (2 x 10 L). The combined organic phase was washed with sat.Na2SO3 (2 x 8 L), brine (8 L), and dried over Na2SO4. The organic phase was concentrated under vacuum. The residue was slurried in MeOH (4 L), filtered, and dried to offer 3-chloro-2-fluoro-4-iodopyridine 1c (2.2 kg, yield 68%).
Step b
[00311] Into a solution of Compound 1c (8 kg) in DMSO (48 L) was passed through NH3
(gas) at 80 °C overnight. TLC showed the reaction was finished. The reaction mixture was cooled to RT. The reaction mixture was added to water (140 L). The solid was collected and washed with water (25 L), dried to afford Z17b (6.91 kg, yield 87%). 1H NMR (400 MHz, CDC13) δ = 7.61 (d, J= 6.8 Hz,
1H), 7.14 (s , J= 6.8 Hz, 1H), 5.09 (bs, 2H).
Step c
[00312] A solution of 2-amino-6-chloro-pyrazine la (1 kg, 7.69 mol) in DCM (15 L) was heated to reflux, to which was charged NBS (4l7g) in portions during 1 h. The reaction was cooled to room temperature. The reaction mixture was washed with water (3 L) and brine (3 L). The organic phase was evaporated, and the residue was purified by column chromatography to give product Z17f
[00313] To a solution of 3-bromo-6-chloropyrazin-2-amine Z17f (6.0 kg, 28.78 mol) in 1,4- Dioxane (40 L) was added Pd(OAc)2 (64.56 g, 287.6 mmol), Xantphos (333 g, 575.6 mmol), and DIPEA (7.44 kg, 57.56 mol) at room temperature under nitrogen. After another 30 minutes purging with nitrogen, methyl 3-mercaptopropanoate (3.81 kg, 31.70 mol) was added, resulting in darkening of the orange mixture. The mixture was heated to 90°C. HPLC showed complete conversion of the starting material. The mixture was allowed to cool to about room temperature, then diluted with EtOAc (40L). After aging for 30 min with stirring, the entire mixture was filtered and solids were washed with EtOAc (3 x 15L). The combined orange filtrate was concentrated to dryness and the solid residue was suspended in DCM (45 L). The mixture was heated to 35-40 °C and stirred for 1h until all solids were dissolved. Then n-heptane (45L) was added dropwise. Upon complete addition, the mixture was cooled to 15-20 °C with stirring for 1h. The solids were collected by vacuum filtration and solids were washed with cold 1:1 DCM/heptane (25 L), then heptane (25 L) (PSC-2). The solids were dried over the weekend to give Z17d (5.32 kg, yield 75%). 1H NMR (400 MHz, CDCl3) δ = 7.83 (s, 1H), 4.88 (bs,
[00314] To a solution of Z17d (8.0 kg, assay 95%, 30.68 mol) in THF (70 L) was added
EtONa (prepared from 776 g Na and 13.6 L EtOH) at room temperature and the mixture was stirred at
ambient temperature for 1 hour. The mixture was then concentrated to a wet yellow solid by rotary evaporation and the residue was suspended in DCM (40L). The mixture stirred under N2 for l6h. The solids were collected by vacuum filtration and the cake was washed with DCM (about 15 L) until the filtrate was colorless (PSC-2). The solids were then dried under vacuum to give Z17c (6.93 kg, qNMR
[00315] To a mixture of Z17c (6.95 kg, assay 72%, 27.23 mol) in l,4-dioxane (72 L) was added Xantphos (233 g, 411 mmol, 0.015 eq), Pd2(dba)3 (186 g, 206 mmol, 0.0075 eq), Z17b (7.13 kg, 28.02 mol) and DIPEA (7.02 kg, 54.46 mol). The system was vacuated and purged with nitrogen gas three times. The mixture was stirred at 65 °C for 16 h under N2. The mixture was cooled to RT and water (50 L) was added, filtered. The cake was washed with EA (25 L). The filtrate was extracted with EA (4 x 20 L). The organic phase was concentrated in vacuum to offer the crude product which was combined with the cake. Then DCM (60 L) was added to the crude product and stirred at 25-30°C for l8h and then filtered. The filter cake was slurried with CH2Cl2 (30 L) for 4 hrs and filtered. The filter cake was slurred in CH2Cl2 (30 L) for 16 hrs and filtered. Then the filter cake was dried in vacuum to give Z17a (3-((2-amino-3-chloropyridin-4-yl)thio)-6-chloropyrazin-2-amine; 9.1 kg, 84 %) as light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ = 7.89 (s, 1H), 7.7 (d, J= 7.6 Hz, 1H), 7.18 (bs, 2H), 6.40 (bs, 2H), 5.97 (d, J= 7.6 Hz, 1H).
Example 4
Alternative formation of the intermediate Z17a (here also named Y7a)
[00316] By way of alternative and according to a preferred reaction method, the compound of the formula Z17a was obtained according to the following Reaction Scheme:
In detail, the synthesis of the compound of the formula Y7a = Z17a was carried out as follows:
(25% wt, 364.00 g, 400 mL, 2.68 mol, 6.14 equiv) were added to a 1-L sealed reactor. The mixture was heated to 80 °C and stirred for 24 h, and the reaction was completed. The reaction mixture was cooled to 30 °C and filtered to give a brown filter cake. The brown filter cake was dissolved in acetone
(50 mL), and filtered. To the filtrate was added petroleum ether (300 mL). The suspension was stirred for 4 h, and filtered to give the crude product. The crude product was slurried in combined solvents of petroleum ether and acetone (10/1, 200 mL) and filtered to give the product Y7d (51.00 g, 307.91 mmol, 80% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ = 7.63 (s, 1H).
Step b
[00318] To a 200 mL round bottom flask was added Na2S (10.816 g, 44wt% containing crystalline water, 60.978mmol) and toluene (100 mL). The mixture was heated to reflux, and water was removed with a Dean-Stark trap (about 5~6 mL water was distilled out). After cooling, the mixture was concentrated to dryness.
[00319] To above round bottom flask was added Y7d (5.000 g, 30.489mmol) and 2-methylbutan-2-ol (50 mL), the reaction was heated to reflux and stirred for 36 h. After cooling to 25 °C, the mixture was filtered. The solvent of the filtrate was exchanged with n-heptane (5 V, 3 times, based on Y7d), and finally concentrated to IV residue. THF (25 mL) was charged to the residue at 25 °C and stirred. The suspension was filtered and washed with THF/n-heptane (5 mL/5 mL) to give a brown solid (6.200 g).
[00320] To another 200 mL round bottom flask was added above brown solid (6.200 g),
10% brine (25 mL), Me-THF (30 mL) and n-Bu4NBr (9.829 g, 30.489 mmol). The mixture was stirred for 0.5 h at room temperature, and the phases were separated. The organic phase was washed with 20% brine (25 mL), and exchanged the solvent with iso-propanol (5 V *3 times, based on Y7d) to give the iso-propanol solution of Y7c (27.000g, 99.2% purity by HPLC area, 58.08% assay yield). 1H NMR (400 MHz, DMSO-d6) δ = 6.88 (s, 1H), 2.97 – 2.92 (m, 14H), 1.38 – 1.31 (m, 14H), 1.13 – 1.04 (m,
14H), 0.73 – 0.69 (t, 21H).
Step c
[00321] To a 25-mL round-bottom flask was added Y7c (4.7g, 23.27wt%, IPA solution from Step b, 2.723 mmol, 1.0 equiv), Y7b (1.052 g, 4.085 mmol, 1.5 equiv), l,lO-Phenanthroline (0.05 g, 0.272 mmol) and water (8 mL). The mixture was purged with nitrogen gas three times, and Cul (0.026 g, 0.136 mmol) was added under nitrogen atmosphere. The mixture was heated up to 65 °C and stirred for 3 h, and the reaction was completed. The reaction was cooled to room temperature and filtered, and the filter cake was washed with water (4 mL*3). The filter cake was slurried in MTBE (6 mL) for 30 min and filtered. The filter cake was washed with MTBE (6 mL) and dried to afford Y7a which is Z17a (565 mg, 72% yield).
[00322] Z17b is synthesized as described in Example 3 Step a and Step b.
Example 5
Alternative Synthesis of the intermediate Z17a:
[00323] According to another preferred method, the compound of the formula Z17a was obtained in accordance with the following Reaction Scheme:
[00324] The reactions were carried out as follows:
Step a
Y7d was synthesised as described in Example 4 step a.
Step b
[00325] To a three-necked round-bottle flask was added Y7d (200 mg, 1.22 mmol, 1 equiv), dioxane (4 mL). The solution was vacuated and purged with nitrogen gas three times. Xantphos (14mg, 0.024 mmol, 0.02 equiv), PdCl2(dppf) (8.9 mg, 0.012 mmol, 0.1 equiv), and DIPEA (0.32 g, 2.44 mmol, 2.0 equiv) were added under nitrogen atmosphere. The solution was heated to 85 °C for overnight. The reaction was cooled and evaporated. The residue was purified by column chromatography (eluent/ethyl acetate/heptane = 1/1) to give Z17d (259 mg, 0.99 mmol, 81%). 1H NMR (400 MHz, CDCl3) δ = 7.83 (s, 1H), 4.88 (bs, 2H), 3.73 (s, 3H), 3.47 (t, J= 9.2 Hz, 2H), 2.79 (t, J= 9.2 Hz, 2H).
[00326] The remaining steps were carried out as described in Example 4, Steps e and f, to yield Z17a. Z17b was synthesized as described in Example 3 Step a and Step b.
Example 6
(3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8- azaspiro[4.5]decan-4-amine. succinate (1:1) hemihydrate. modification (form) HA:Variant a)
[00327] 50 ml ethanol and 2.5 ml water were added to a 100ml flask containing 3.0 g of free base of 3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine (obtained as A18 for example as described in Example 1) and 848.0 mg of succinic acid. The mixture was heated to 50°C to generate a clear solution. The temperature was lowered to 15°C during a period of 3 hours. The solution was kept stirring at 15°C overnight.
Precipitated solid was separated via suction filtration and 50 ml of acetone was added to produce a suspension. The suspension was stirred at 50°C for 3 hours. The solid was separated with suction filtration and dried at room temperature under vacuum for 3 hours. Yield was about 60%.
[00328] The succinate appeared as a highly crystalline solid, with a melting point onset of
94.4°C and an accompanying enthalpy of 96 J/g. The succinate salt crystals showed aggregates of broken drusy tabular particles.
[00329] Variant b)
[00330] 14.34 g of 3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)- 3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine free form (obtained as A18 for example as described in Example 1) and 4.053 g of succinic acid were equilibrated in 100 mL 95% EtOH at 50°C. Add 5 mL of water into the system and heat to 70-75 °C. Add 95 mL of pure EtOH and heat for 30 min more. Stir over night at 25 oC. Filter the mixture wash with EtOH and dry under vacuum in an oven at room temperature. Yield is 87.5%.
SHP2 is a nonreceptor protein tyrosine phosphatase encoded by the PTPN11 gene and is involved in cell growth and differentiation via the MAPK signaling pathway. SHP2 also plays an important role in the programed cell death pathway (PD-1/PD-L1). As an oncoprotein as well as a potential immunomodulator, controlling SHP2 activity is of high therapeutic interest. As part of our comprehensive program targeting SHP2, we identified multiple allosteric binding modes of inhibition and optimized numerous chemical scaffolds in parallel. In this drug annotation report, we detail the identification and optimization of the pyrazine class of allosteric SHP2 inhibitors. Structure and property based drug design enabled the identification of protein–ligand interactions, potent cellular inhibition, control of physicochemical, pharmaceutical and selectivity properties, and potent in vivo antitumor activity. These studies culminated in the discovery of TNO155, (3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine (1), a highly potent, selective, orally efficacious, and first-in-class SHP2 inhibitor currently in clinical trials for cancer.
Step a: A mixture of (3S,4S)-tert-butyl 4-((R)-1,1-dimethylethylsulfinamido)-3-methyl-2-oxa-8- azaspiro[4.5]decane-8-carboxylate (51 mg, 0.136 mmol) and HCl (4 M in dioxane, 340 L, 1.362 mmol) in MeOH (5 mL) was stirred for 1 h at 40 °C. After cooling to RT, the volatiles were removed under reduced pressure to give (3S,4S)-3-methyl-2-oxa-8-azaspiro[4.5]decane-4-amine which was used in next step without further purification. MS m/z 171.1 (M+H)+. Step b: A mixture of (3S,4S)-3-methyl-2-oxa-8-azaspiro[4.5]decane-4-amine crude, 3-((2-amino3-chloropyridin-4-yl)thio)-6-chloropyrazin-2-amine (35.5 mg, 0.123 mmol), and DIPEA (193 L, 1.11 mmol) in DMSO (600 L) was stirred for 16 h at 100 °C. After cooling to RT, the volatiles were removed under reduced pressure and the resulting residue was purified by HPLC (gradient elution 15-40% acetonitrile in water, 5 mM NH4OH modifier) to give (3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine (11 mg, 0.026 mmol). 1 H NMR (400 MHz, METHANOL-d4) δ ppm 7.67-7.47 (m, 2 H), 5.91 (d, J=5.5 Hz, 1 H), 4.22 (qd, J=6.4, 4.8 Hz, 1 H), 4.03 (ddt, J=13.5, 8.9, 4.7 Hz, 2 H), 3.86 (d, J=8.7 Hz, 1 H), 3.71 (d, J=8.7 Hz, 1 H), 3.37 (td, J=9.9, 4.9 Hz, 1 H), 3.29-3.23 (m, 1 H), 3.00 (d, J=5.0 Hz, 1H) 1.91-1.56 (m, 4 H), 1.21 (d, J=6.4 Hz, 3 H). HRMS calcd for C18H25ClN7OS (M+H)+ 422.1530, found 422.1514.












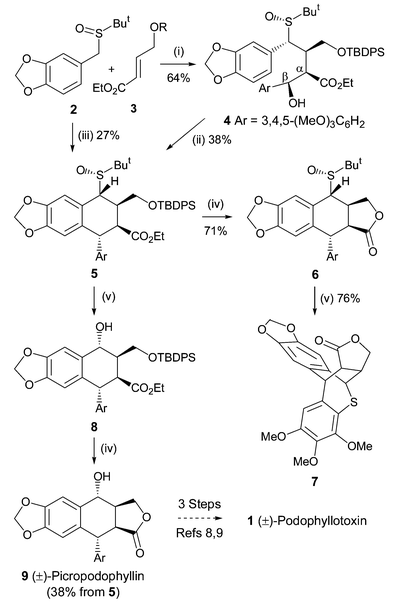
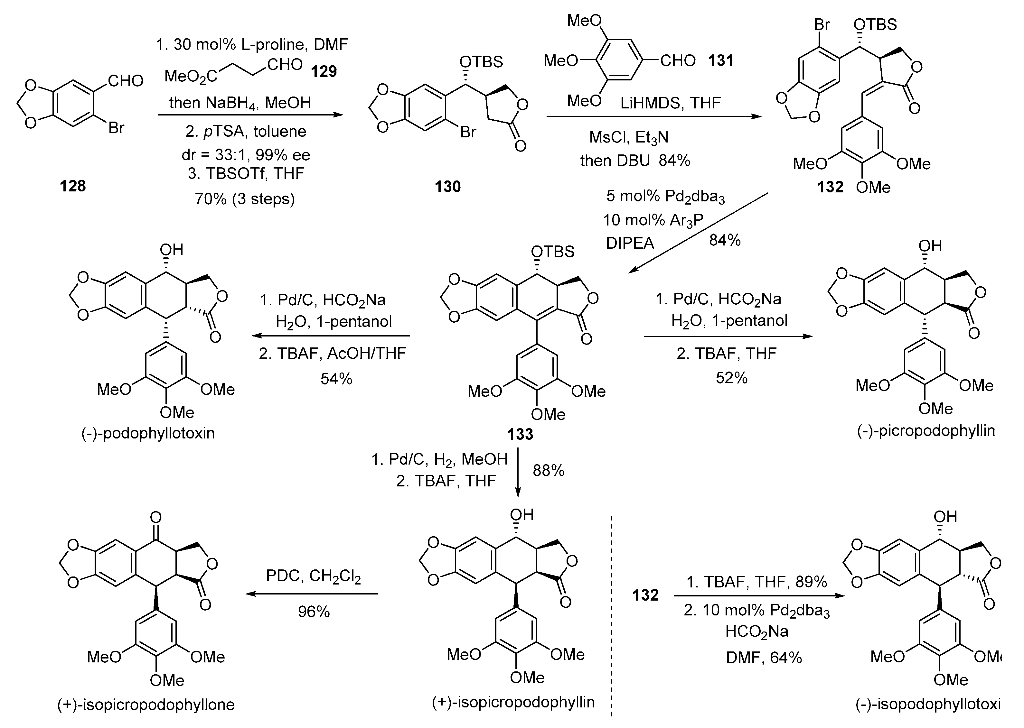






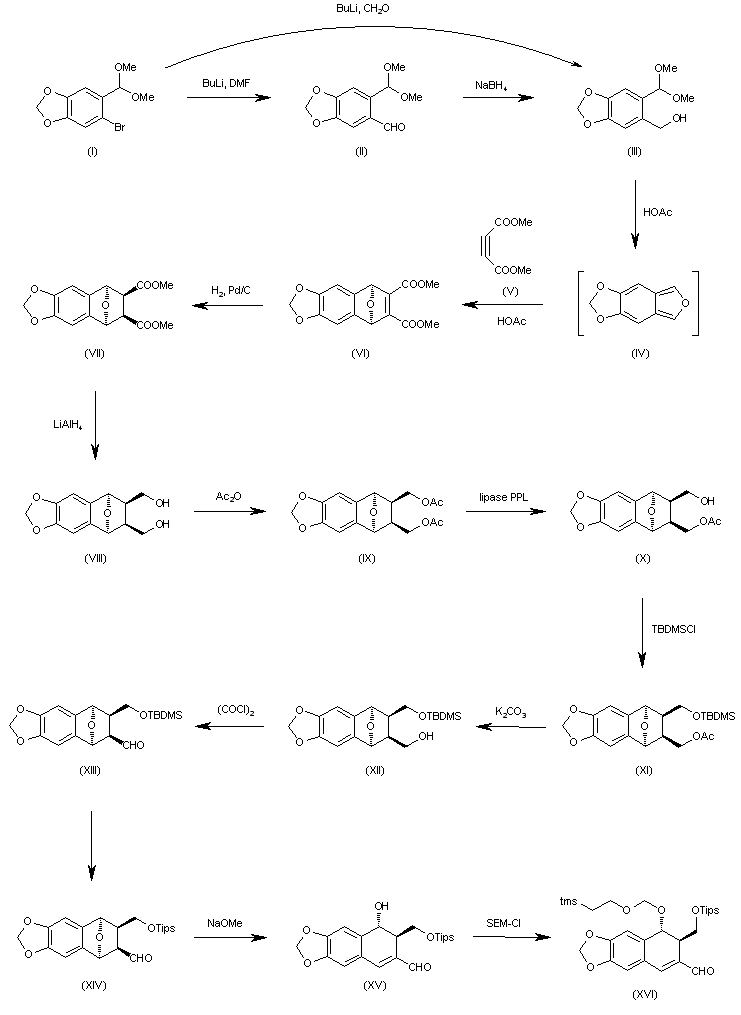
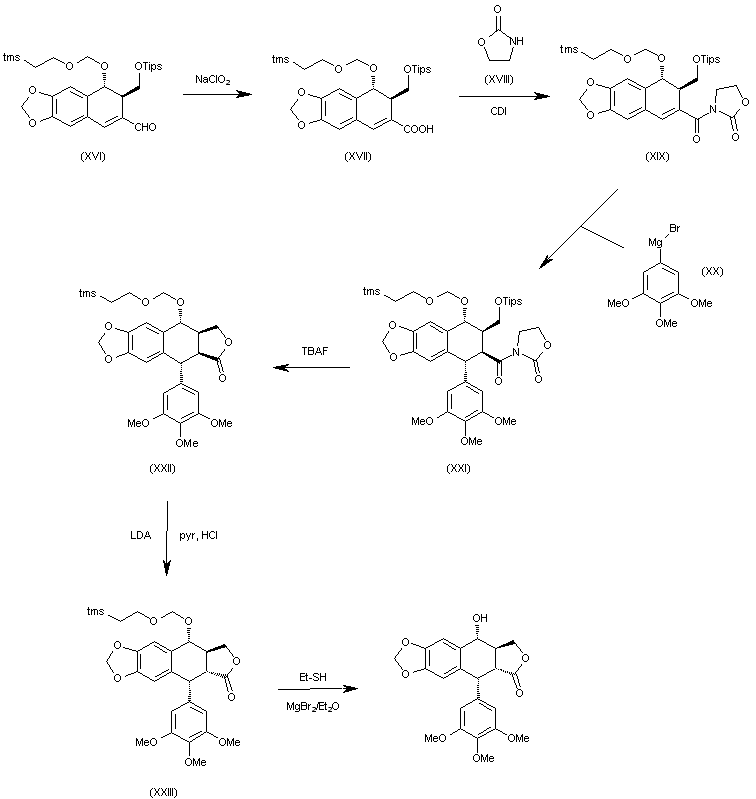
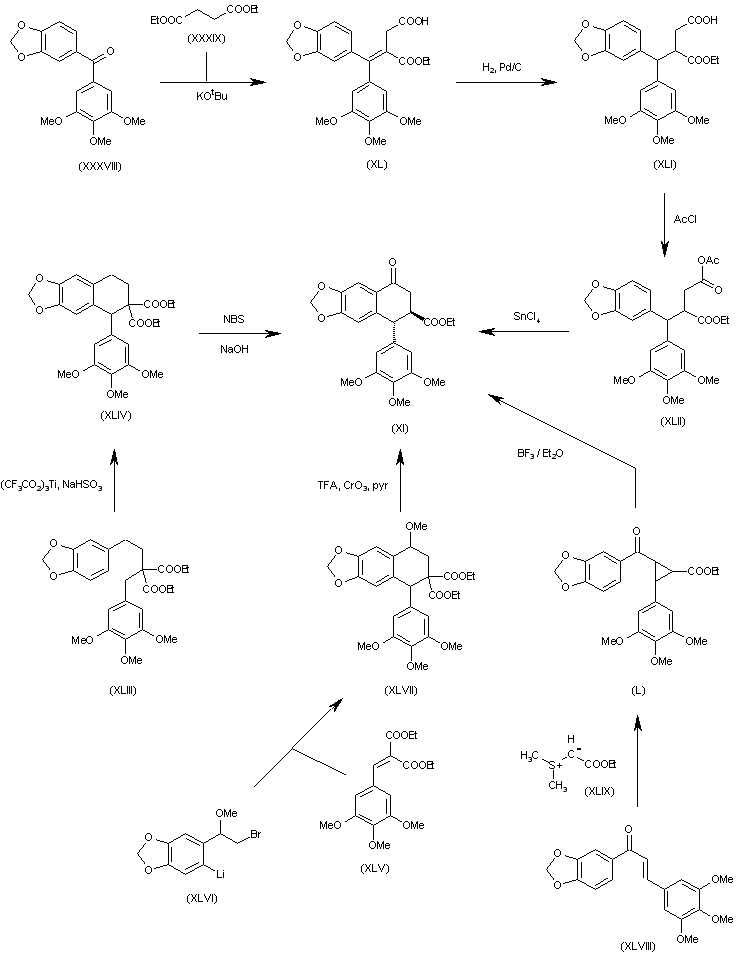
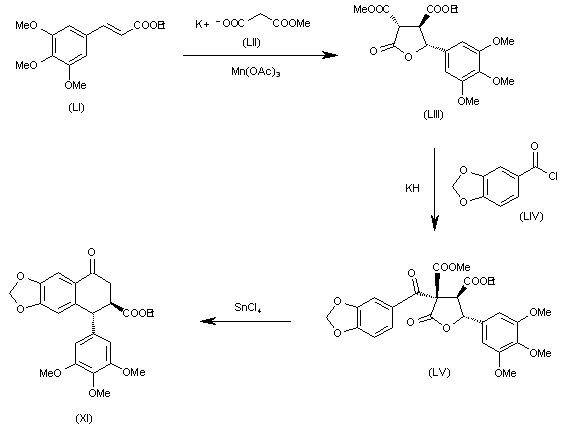
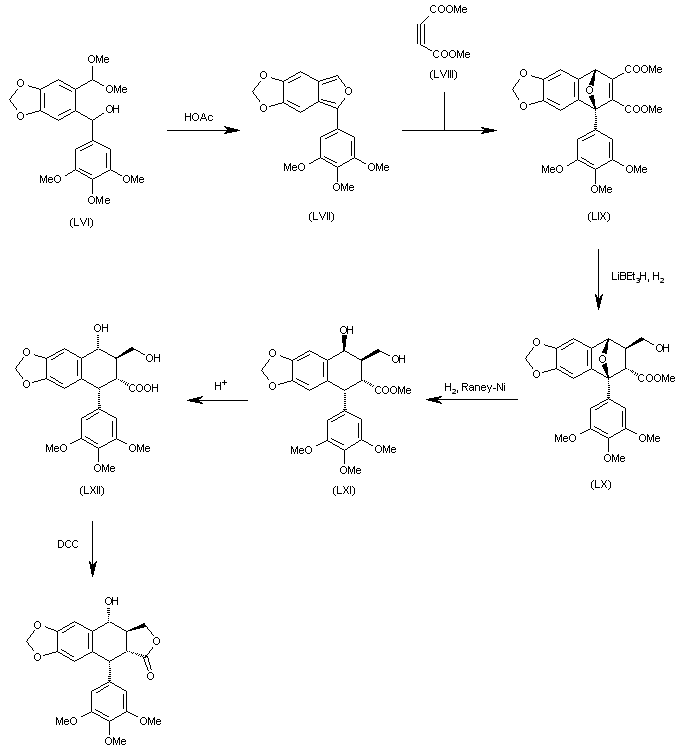
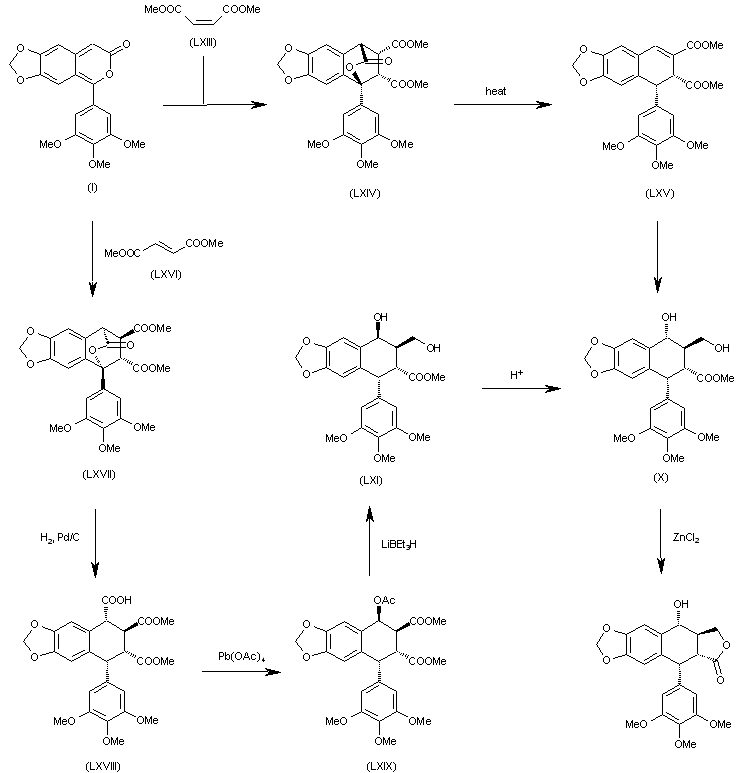
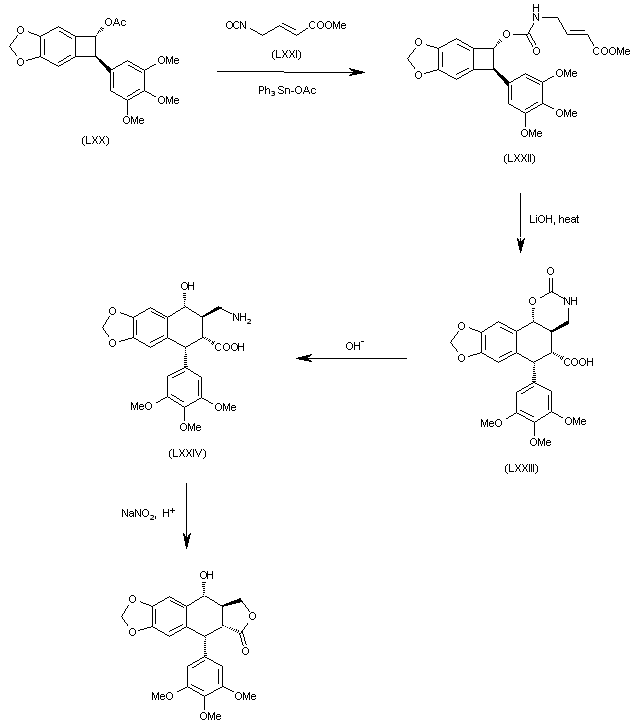
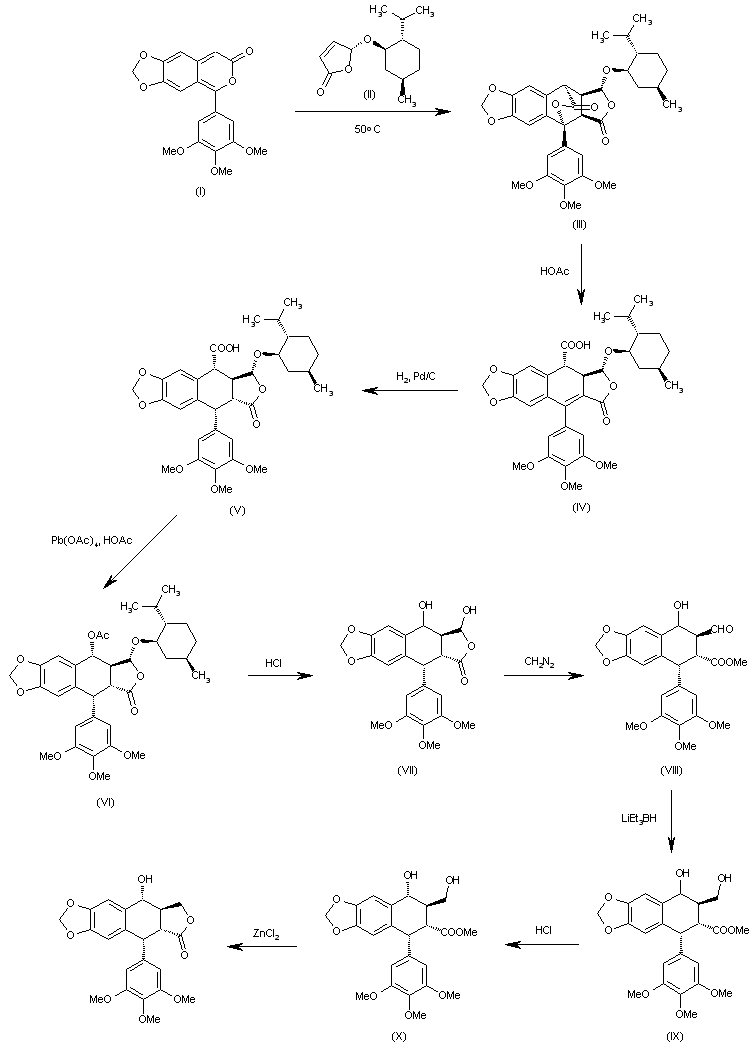







 compound. Selinexor functions by binding with and inhibiting the nuclear export protein XP01 (also called CRM1 ), leading to the accumulation of tumor suppressor proteins in the cell nucleus. This reinitiates and amplifies their tumor suppressor function and is believed to lead to the selective induction of apoptosis in cancer cells, while largely sparing normal cells. Over 1 ,200 patients have been treated with Selinexor in company and investigator-sponsored Phase 1 and Phase 2 clinical trials in advanced hematologic malignancies and solid tumors. Karyopharm has initiated four later-phase clinical trials of Selinexor, including one in older patients with acute myeloid leukemia (SOPRA), one in patients with Richter’s transformation (SIRRT), one in patients with diffuse large B-cell lymphoma (SADAL) and a single-arm trial of Selinexor and lose-dose dexamethasone in patients with multiple myeloma (STORM). Patients may receive a twice-weekly combination of Selinexor in combination with low dose dexamethasone. Randomized 1 :1 , Selinexor will be dosed either at 60mg + dexamethasone or at 100 mg + dexamethasone.
compound. Selinexor functions by binding with and inhibiting the nuclear export protein XP01 (also called CRM1 ), leading to the accumulation of tumor suppressor proteins in the cell nucleus. This reinitiates and amplifies their tumor suppressor function and is believed to lead to the selective induction of apoptosis in cancer cells, while largely sparing normal cells. Over 1 ,200 patients have been treated with Selinexor in company and investigator-sponsored Phase 1 and Phase 2 clinical trials in advanced hematologic malignancies and solid tumors. Karyopharm has initiated four later-phase clinical trials of Selinexor, including one in older patients with acute myeloid leukemia (SOPRA), one in patients with Richter’s transformation (SIRRT), one in patients with diffuse large B-cell lymphoma (SADAL) and a single-arm trial of Selinexor and lose-dose dexamethasone in patients with multiple myeloma (STORM). Patients may receive a twice-weekly combination of Selinexor in combination with low dose dexamethasone. Randomized 1 :1 , Selinexor will be dosed either at 60mg + dexamethasone or at 100 mg + dexamethasone.





![2-(4-Chlorophenyl)-N-[[2-(2,6-dioxopiperidin-3-yl)-1-oxo-3H-isoindol-5-yl]methyl]-2,2-difluoroacetamide.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=118647211&t=l)








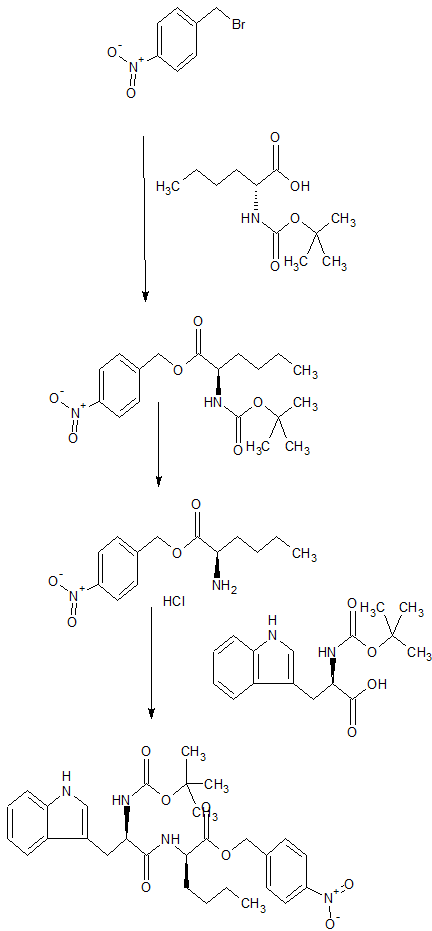




![N-[3-[2-[2,3-Difluoro-4-[4-(2-hydroxyethyl)piperazin-1-yl]anilino]quinazolin-8-yl]phenyl]prop-2-enamide.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=117909640&t=l)




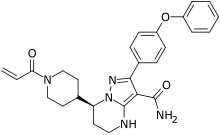




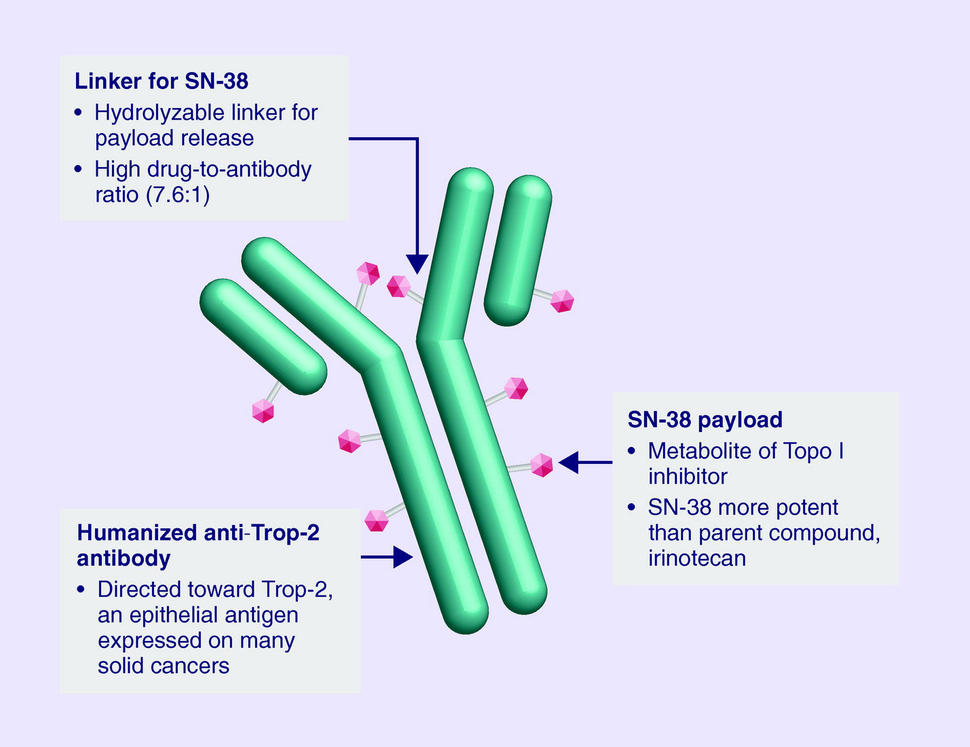
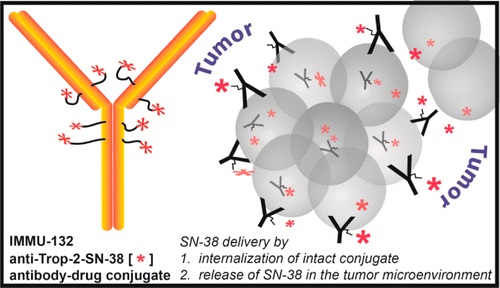







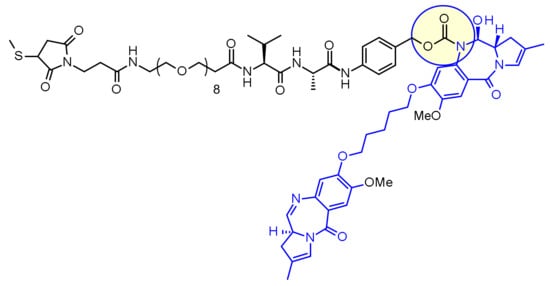


![4-((S)-4-Acryloyl-2-methylpiperazin-1-yl)-6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=137278711&t=l)
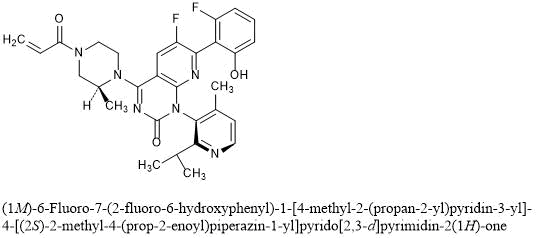






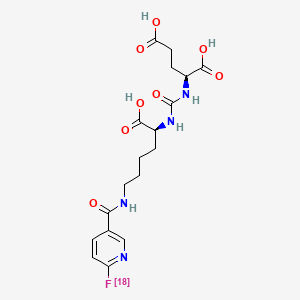
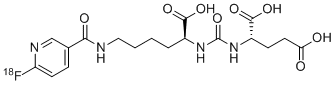


![Radiosynthesis of [ 18 F]DCFPyL](http://www.researchgate.net/publication/301830909/figure/fig4/AS:362696278069251@1463484939571/Radiosynthesis-of-18-FDCFPyL.png)

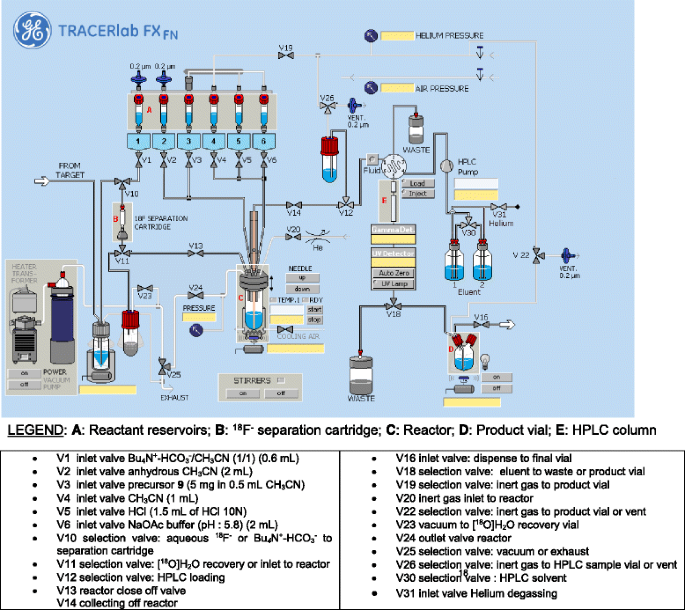
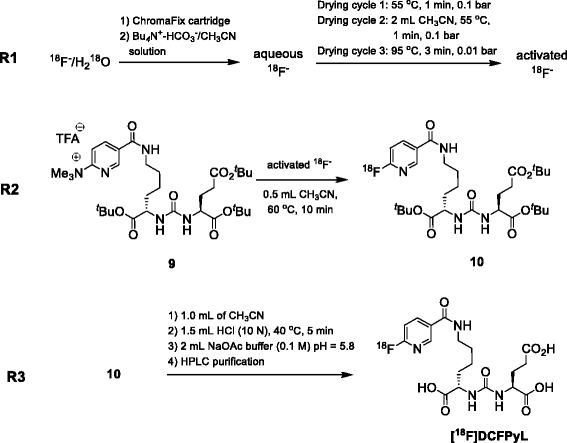
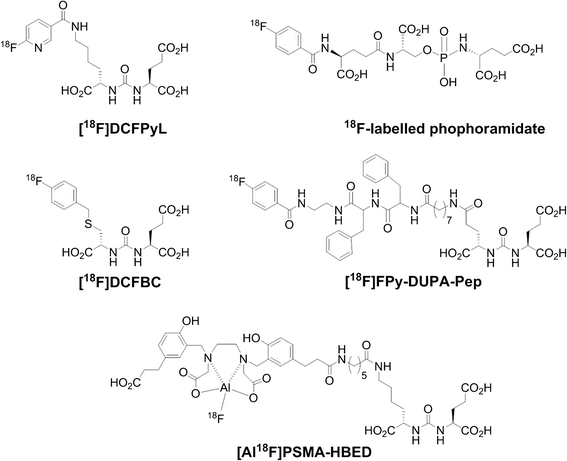


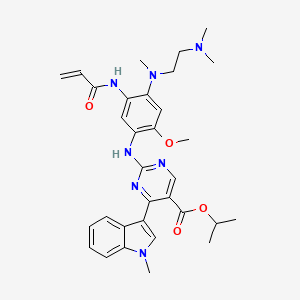

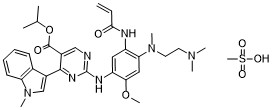
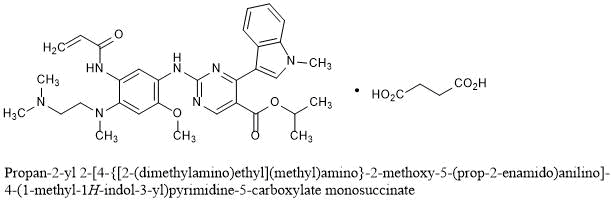
 C4H6O4 : 703.78
C4H6O4 : 703.78















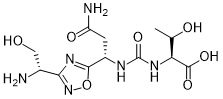


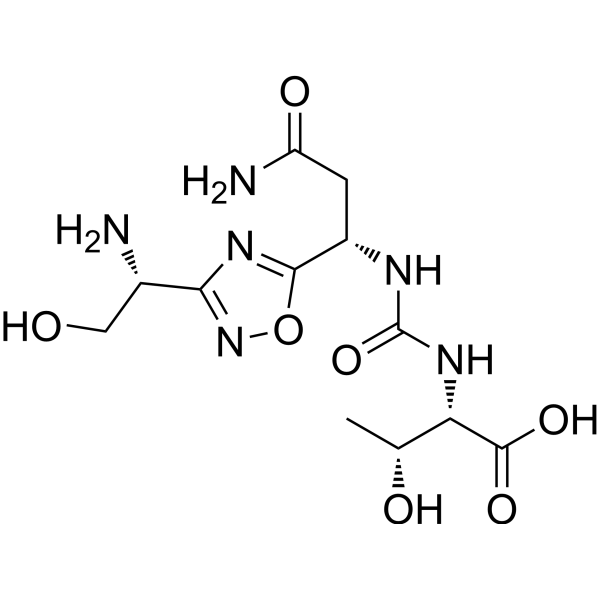



 Jan 21, 2015
Jan 21, 2015